-Immunologie
-Herz-Kreislauf
-Pathophysiologie d. Niere
-Krebsentstehung
-Knochenstoffwechsel
-Muskelfunktionsstörungen
-Gicht
INHALT:
1. Steinzeit-Anpassung
2. Überernährung
- Insulinresistenz
- FTO-Gen, braunes Fett
- Einfluss der Mikrobiota
- Tipps zum Abnehmen
3. Hohe Salzaufnahme
4. Unterernährung
- Essstörungen
- Ressourcenknappheit
- Kwashiorkor
5. Appetit-Regulation
- Portionsgrößeneffekt
- Leptin
- Hunger
- PYY
- GLP-1
- Ghrelin
6. Kohlenhydrat-Verdau
- Glykämischer
Index
- Laktose-Intoleranz
- Fructosemalabsorption
- Fructose-Kontroverse
7. Eisen
- Hämochromatose
8. Folsäure
9. Vitamin B12
- Perniziöse
Anämie
10. Selen
11. Fettsäuren
- Omega-3-Fettsäuren
- Trans-Fettsäuren
12. Proteine, Magen
13. Ulcus
14. Protein-Verdauung
15. Pankreatitis
16. Zystische Fibrose
17. Zöliakie
18. Inflammatory bowel d.
19. Leber
Energiestoffwechsel
- Resorptionsphase
- Postresorptionsphase
- Hungerphase
- Ketogene Diät
- Diabetes Mellitus Typ 1
- Stg. Gluconeogenese
- Metab. Syndrom
- Metformin
Aminosäure-Stoffwechsel, Harnstoffsynthese
- Säure-Basen-Regulation
Partikelfilter
Biotransformation
- Cytochrom Oxidasen
- CYP-Induktion
- Xenohormone
- Wechselwirkungen
- -Alkohol-Abbau
- -Paracetamol
Steroid-Inaktivierung
Bilirubin
Cholesterin
Cholestase
- Gallensäuren
- Gallensteine
Fettverdauung
Plasmaproteine
Portale Hypertension Hepatorenales Syndrom
SKRIPT ERNÄHRUNG UND VERDAUUNG
Arno Helmberg
Dieses Skriptum ist eine Lernhilfe zu meiner Vorlesung im Modul "Ernährung und Verdauung" an der Medizinischen Universität Innsbruck. Es steht auch in einer pdf-Version sowie in einer Englischen Version zur Verfügung.
Version 6.3 ©Arno Helmberg 2000
Einfach ausgedrückt, ist die Strategie
folgende: komplexe und damit risikoreiche Substanzen bleiben draußen.
Dort werden sie in allgemein akzeptierte, sichere Bestandteile zerlegt.
Nur diese werden über definierte Eintrittspforten importiert. Allerdings
müssen wir zu dieser Politik Ausnahmen machen. Einerseits benötigen
wir einige hochkomplexe Moleküle, die wir nicht selbst herstellen können,
z. B. Vitamin B12. Um diese vor Verdauung zu schützen und zu importieren,
brauchen wir spezielle Dienste, so wie es Spezialisten benötigt, um alte Gemälde unbeschadet
über geographische und politische Barrieren zu transportieren. Andererseits müssen
wir manche in den Darm sezernierten Substanzen in großen Mengen recyceln und setzen
dazu, z. B. wegen der Vielgestaltigkeit von Gallensäuren und –salzen, relativ
unspezifische Transportmechanismen ein, durch die manchmal auch gefährliches
Material aufgenommen wird. Schließlich müssen wir, gerade um unsere Grenzen zu
schützen, unserem Verteidigungssystem die Gelegenheit geben, sich mit den
potentiellen Gefahren jenseits der Grenzen auseinanderzusetzen und diese von
den zahlreichen harmlosen Nahrungsmittel-antigenen und gutartigen bakteriellen
Symbionten unterscheiden zu lernen, die unser Organismus tolerieren soll. Zu
diesem Zweck öffnen wir Kanäle via M-Zellen, über die wir kleine Stichproben
komplexer Strukturen aufnehmen. Obwohl wir diese Kanäle sorgfältig überwachen,
können sie manchmal dazu missbraucht werden, pathogene blinde Passagiere einzuschleusen.
1. EVOLUTIONÄRE GESICHTSPUNKTE
Was ist gesunde Ernährung? Diese
Frage berührt jeden und löst manchmal heftige Auseinandersetzungen aus. Anhänger
heißer Ernährungstrends bekämpfen einander mit Gusto. Der in reichen Ländern
vielfach praktizierte tägliche Genuss von fettreichen Fleischprodukten, häufig
stark gesalzen oder geräuchert als Wurst oder Speck, ist definitiv nicht
gesund. Jedoch auch das Gegenteil, eine rein vegetarische oder vegane
Ernährung, stellt eine Mangelernährung dar. Dabei muss zumindest Vitamin B12 supplementiert
werden; darüber hinaus gibt es eine Reihe von potentiell kritischen Nährstoffen,
besonders in bestimmten Lebenssituationen erhöhten Bedarfs: bei Babys, Kindern,
Jugendlichen, Schwangeren und Stillenden. Dazu gehören Protein bezüglich Menge
und Aminosäurenzusammensetzung, langkettige n‑3-Fettsäuren, Eisen, Zink, Vitamin
D, Calcium und Jod. Nicht selten finden sich solche Mangelerscheinungen bei
Kindern vegan lebender Eltern: In der besten Absicht gehen diese davon aus,
dass die Ernährungsform, die sich für sie jahrelang bewährt hat, auch für die
Kinder gut sein muss. Wenn auf potentiell kritische Nährstoffe geachtet wird
und diese bei Bedarf supplementiert werden, ist eine vegetarische lebensweise
durchaus gesund: Vegetarier haben weniger Herzinfarkte, Schlaganfälle und seltener
Diabetes mellitus Typ 2, hohen Blutdruck sowie manche Krebsarten.
Um die Anforderungen unseres Organismus
zu verstehen, hilft ein Blick in unsere Geschichte. Während der moderne
Homo sapiens über die ganze
Welt verbreitet lebt, ist das eine relativ neue Entwicklung. Aus Analysen
von mitochondrialer DNA sowie des Y‑Chromosoms wissen wir, dass
alle aus Europa, Asien, Australien und den amerikanischen Kontinenten
stammende Menschen die Nachfahren einer relativ kleinen Gruppe von Emigranten
darstellen, die vor 60.000 bis 70.000 Jahren Afrika und den Nahen Osten
verließen. In evolutionären Maßstäben gemessen ist diese Zeit zu kurz
für große genetische Anpassungsmechanismen. Mit anderen Worten, wir
sind zu einem großen Ausmaß das Produkt von Selektionsmechanismen, die
im Paläolithikum in Afrika herrschten. Wir sind auf Bedingungen der
Altsteinzeit optimiert.
Leider ist es unmöglich, die Speisekarte dieser
Zeit genau zu rekonstruieren, doch sind einige Eckpunkte sehr wahrscheinlich.
Nahrung war oft knapp. Der Selektionsdruck favorisierte
jene Individuen, die in der Lage waren, bei gelegentlich guter Versorgungslage
effizient Fettreserven aufzubauen, um die langen Hungerperioden zu überstehen:
Leute mit thrifty genes, sparsamen
Allelen. Das fettarme Fleisch erlegten Wildes war begehrt, aber schwer zu erjagen.
Für einen großen Teil der Ernährung war man daher auf Pflanzen angewiesen,
Wurzeln, Knollen, Nüsse und in feuchteren Regionen, Früchte. Der
Kohlenhydratanteil der Ernährung war generell geringer. Kohlenhydrate waren
überwiegend komplexe Mischungen aus Stärke und unverdaulichen Fasern.
"Süße" Einfachzucker wie Glucose oder Fructose gab es selten, z. B.
in Beeren im Herbst oder in wildem Honig. Milch und Getreide, Produkte der
neolithischen (Ackerbau‑) Revolution vor 10.000 Jahren, gab es noch nicht.
Im Vergleich zu unserer durchschnittlichen Westlichen Ernährung enthielt die paläolithische
Ernährung also wahrscheinlich mehr Protein und Ballaststoffe, mäßig Fett und
weniger, vor allem weit weniger kurzkettige, Kohlenhydrate. Ausreichend Nahrung
zu sammeln bedeutete tägliches Wandern und harte körperliche Arbeit. Um in der
afrikanischen Sonne nicht auszutrocknen, war es für den Organismus notwendig,
effizient Wasser zu sparen. Aus osmotischen Gründen bedeutete das auch, Salz zu
sparen, das, außer an der Küste, knapp war.
Archäologische Funde belegen, dass die
neolithische Revolution einen negativen Einfluss auf die Gesundheit des
Individuums hatte. Die frühen Ackerbauern litten an Mangelernährung, waren kleiner,
hatten schlechte Zähne und eine niedrigere Lebenserwartung.
Mit diesen Überlegungen möchte ich Bedingungen verständlich machen, die für unsere Ernährung und Gesundheit grundlegend sind. Ich möchte damit nicht suggerieren, dass wir uns wie in der Altsteinzeit ernähren sollten, obwohl ich glaube, dass eine solche Ernährung gesund wäre. Erstens ist das für die meisten von uns unmöglich. Das Fleisch der Altsteinzeit war etwas qualitativ anderes als das uns zur Verfügung stehende: Fleisch von Wild enthält wesentlich weniger Fett als das unserer Masttiere. Zweitens sind Getreideprodukte, Mais und Reis angesichts der Menge der benötigten Kalorien nicht zu ersetzen. Wir müssen uns bei einer Weltbevölkerung von acht Milliarden Menschen anders verhalten als bei einer altsteinzeitlichen Zahl von höchstens einigen hunderttausend. Die Massentierhaltung, die nötig wäre, um so viele Menschen häufig mit Fleisch zu versorgen, ist ethisch und nach Klimaerwägungen wohl nicht vertretbar.
Der Ernährung unserer altsteinzeitlichen afrikanischen Vorfahren am nächsten kommt heute wahrscheinlich die "Mediterrane Ernährung". Zum Bedauern (wahrscheinlich nicht nur) des Schreibers dieser Zeilen ist damit aber nicht Pasta, Lasagne und Crème brûlée gemeint. Bei Untersuchungen nach dem 2. Weltkrieg fiel auf, dass die Menschen auf Kreta besonders gesund waren und besonders alt wurden. Diese rurale, körperlich hart arbeitende, gewiss nicht wohlhabende Population ernährte sich mit viel Gemüse, Salaten, Obst, Fisch und Olivenöl und mit wenig Hühner- und Schaffleisch sowie wenig Zucker.
Wir werden einen Kompromiss zwischen der biologisch optimalen
und der ethisch wünschenswerten Ernährung finden müssen, der zudem auf der
globalen Ebene wirtschaftlich machbar und fair ist. Doch ist das nicht das
Thema dieser Vorlesung. Mein Ziel hier ist es, die gesundheitlich relevanten
Aspekte der Ernährung verständlich zu machen.
2. ÜBERERNÄHRUNG
Für Menschen, die heute in reich versorgten
Ländern leben, ist die genetische Ausstattung der Altsteinzeit nicht optimal. Durch
unseren bewegungsarmen Tagesablauf wird das Missverhältnis noch verstärkt. Was
das Nahrungsangebot betrifft, leben wir in einem Dauerfest. Stellen wir uns dem
nicht bewusst entgegen, häufen wir Fettdepots an; eine unserer drückendsten
gesundheitlichen Herausforderungen.
Was ist zu viel? Das Körpergewicht wird am
häufigsten anhand des BMI (body mass
index, Körpergewicht in kg/ Quadrat der Körpergröße in m) eingeordnet: Normalbereich
für Frauen 19-24, für Männer 20-25. Darüber übergewichtig, ab 30 adipös. Der
BMI wird routinemäßig angewendet, da er einfach zu bestimmen ist, sagt aber
nichts über die Verteilung des Gewichts zwischen Fett und lean body mass aus: ein BMI 30 ist bei einer Hammerwerferin anders
zu werten als bei einer Frau, die kaum Sport betreibt. Knochendichte,
Knochendurchmesser, Schulter- und Hüftbreite spielen ebenfalls unberücksichte
Rollen. Auch für große Männer ist ein höherer Wert weniger aussagekräftig.
Einfach und nützlich wegen der Bedeutung des viszeralen Fetts ist auch eine
Bestimmung des Bauchumfangs, für die lediglich ein Maßband benötigt wird. Empfohlen
werden Werte unter 80 cm bei Frauen, unter 94 bei Männern; bei Werten über 88
cm bei Frauen und 102 cm bei Männern besteht ein deutlich erhöhtes
Gesundheitsrisiko. In Bezug gesetzt zur Körpergröße (waist to height ratio) ist er in mancher Hinsicht aussagekräftiger
als der BMI. Für Erwachsene bis 40 Jahre wird ein Wert <0,5 empfohlen, ab 50
Jahren <0,6. Mit nichts anderem als den beiden Werten für Körpergröße und
Bauchumfang kann via einer komplizierteren Formel auch der body roundness index (BRI) berechnet werden, der die
"Fettbelastung" noch besser einschätzen kann (BRI-Calculator: https://webfce.com/bri-calculator/). Eine
apparativ aufwendige, aber aussagekräftigere Bestimmung liefert die
Bioimpedanzanalyse, bei der die Verteilung zwischen Fett- und lean body mass geschätzt und in
Beziehung zu Alter und Körpergröße gesetzt wird. Genauere Methoden zur
Bestimmung des Fettanteils – Unterwasserdensitometrie/air displacement plethysmography, DEXA, 40K+-Zählung,
Magnetresonanztomographie – sind zu aufwändig für den Routineeinsatz.
Die Fettdepots verhalten sich nicht einfach wie
eine inerte Vorratskammer. Je größer diese Depots werden, desto stärker
entwickeln sie eine entzündliche Komponente: Einerseits produzieren diese
Fettzellen selbst TNFα, IL‑6 und das Makrophagen-attrahierende
Chemokin CCL2; andererseits führt das zur Einwanderung und Aktivierung von
Makrophagen, die ihren Cocktail aus IL‑1β, IL‑6 und TNFα
freisetzen. Wir werden sehen, dass sich das negativ auf die Insulinsensitivität
auswirkt. Warum diese entzündliche Komponente? Mehrere Mechanismen werden
diskutiert. Die pralle Füllung der Fettzellen könnte ihre Sauerstoffversorgung
verschlechtern, der so entstandenen Zellstress zur Zytokinproduktion führen.
Einzelne, unzureichend versorgte Adipozyten gehen unter, locken so weitere
Makrophagen an und aktivieren diese. Klar ist jedenfalls, dass mit zunehmendem
viszeralem Fett ein Zustand der niedriggradigen Entzündung im Körper entsteht.
Lokal aktivieren die pro-inflammatorischen Zytokine die Lipolyse, um Energie
zur Bekämpfung des angenommenen Stressauslösers bereitzustellen – es könnte
sich ja um eine Infektion handeln. Das führt zum Anstieg der freien Fettsäuren
im Blut.
Die Akkumulation von Fett beginnt also, toxische
Effekte zu haben. Erinnern wir uns: Toxizität ist eine Frage der Dosis. Vieles deutet
darauf hin, dass der bei Adipositas beobachtete moderate Anstieg der
Konzentration freier Fettsäuren sich auf zwei Ebenen negativ auf den
Metabolismus auswirkt: einerseits auf die Insulin-sensitiven Gewebe,
andererseits auf die Insulin-produzierenden β-Zellen selbst.
Insulinresistenz in Zielorganen: Leber, Fettgewebe, Muskel
Freie Fettsäuren, die nicht zur
Energiegewinnung "verbrannt" werden, akkumulieren im Zytosol von Leber,
Muskel- und Fettgewebe und werden wieder zu Triglyceriden zusammengesetzt.
Dabei steigt auch die Konzentration des Zwischenprodukts Diacylglycerol an, das
sich in die Zellmembran einpflanzt. Erinnern wir uns, dass Diacylglycerol auch als second messenger wirkt, indem es Proteinkinase
C (PKC) an die Membran holt und aktiviert. PKCε phosphoryliert den
Insulinrezeptor auf Threonin 1160 und hemmt damit die
Tyrosinkinasefunktion des Rezeptors, sodass die Signalweiterleitung nicht mehr
so gut funktioniert. In einem weiteren Mechanismus phosphorylieren aktivierte PKC
und andere Serin/Threoninkinasen IRS‑1 (insulin receptor substrate‑1) an hemmenden Stellen und
reduzieren dadurch dessen Fähigkeit, das Insulinsignal in die Muskelzelle zu leiten (nicht verwechseln: im Gegensatz dazu phosphoryliert der aktivierte Insulinrezeptor
IRS‑1 auf Tyrosinen, was zur Signalweiterleitung notwendig ist).
Unabhängig davon sind gesättigte Fettsäuren wie
Laurinsäure (C12) oder Palmitinsäure (C16) in der Lage, TLR4 (den LPS-Rezeptor)
und TLR2 partiell zu aktivieren. Über diesen Mechanismus führen erhöhte Spiegel
freier Fettsäuren zu einer Aktivierung pro-inflammatorischer Signale und zu
weiter reduzierter Insulinsensitivität von Leber, Muskel- und Fettgewebe. So
induzieren aus Fettgewebe freigesetztes IL‑6 und TNFα in Hepatozyten
SOCS (suppressor of cytokine signaling)-Proteine,
die phosphorylierte Tyrosine auf IRS‑1 maskieren, so die
Signalweiterleitung behindern und zum Abbau von IRS‑1 im Proteasom führen.
Regelmäßige aerobe körperliche Betätigung
verstärkt die Expression der für die Fettsäureverbrennung notwendigen Enzyme
und vergrößert die Masse an Mitochondrien, in denen dieser Prozess abläuft. Auf
diese Art führt regelmäßiges Training zu einem effizienteren Abbau von
Fettsäuren und damit zu einer Anhebung der Insulinsensitivität.
Insulinresistenz: β-Zellen im Pankreas
Sobald die peripheren Gewebe weniger sensitiv
auf Insulin sind, ist die Glucosekonzentration im Plasma notwendigerweise
tendenziell erhöht. Glucose wird über den Transporter GLUT2 (KM 15-20 mM) proportional zu seinem Blutplasmaspiegel in die β-Zelle
aufgenommen und stimuliert dort die Produktion von ATP. ATP bindet an einen
ATP-sensitiven K+-Kanal: je höher die ATP-Konzentration, desto mehr
wird der K+-Kanal, der an der Aufrechterhaltung des Membranpotentials
mitwirkt, geschlossen. Das bingt das Membranpotential an die Auslöseschwelle
für spannungsabhängige Ca2+-Kanäle. Einstrom von Ca2+ führt dann zur Exozytose von –Insulin-gefüllten Vesikeln. Mitausgeschüttet wird
ein Proteinhormon namens Amylin oder IAPP (islet
amyloid polypeptide), das gleichsinnig wie Insulin reguliert und gemeinsam
mit Insulin exprimiert und sezerniert wird.
Pharmakologische Querverstrebung:
·
Der ATP-sensitive K+-Kanal (Untereinheit
ABCC8 oder SUR1, sulfonyl urea receptor 1)
ist der Angriffspunkt für Sulfonylharnstoffe. Bindung von Sulfonylharnstoffen
fördert das Schließen des Kanals und verstärkt damit die Insulinausschüttung.
·
Neu begonnene Thiazidtherapie kann zu einem
Anstieg des Blutglucosespiegels führen. Wahrscheinlich ist dieser Effekt auf
die Absenkung des K+-Spiegels zurückzuführen. Extrazelluläre
Absenkung des Kaliumspiegels begünstigt den Ausstrom von K+ aus den
β‑Zellen und behindert so die Insulinfreisetzung.
·
Amylin verzögert die Magenentleerung, verzögert
über mehrere Mechanismen den postprandialen Anstieg von Glucose im Blut und
verringert zentral das Hungergefühl. Ein agonistisches Analogon mit
verlängerter Halbwertszeit, Cagrilintide, wird, in Kombination mit Semaglutid,
bei Patienten mit Diabetes mellitus Typ 2 getestet.
Höhere
Blutzuckerwerte (die Plasmaglucosekonzentration bewegt sich bei Gesunden
zwischen 4 und 8 mM, oder zwischen 72 und 144 mg/dl) führen also zu verstärkter Insulinsynthese und -Freisetzung. Beta-Zellen haben eine enorme Insulinsynthesekapazität:
Proinsulin kann bis zu 50% des gesamten im Pankreas produzierten Proteins
ausmachen. Unter Dauerstimulation führt die Überproduktion an Insulin mit der
Zeit jedoch zu einer Stresssituation im endoplasmatischen Retikulum:
ungefaltetes und fehlgefaltetes Protein akkumuliert. Das betrifft nicht nur Insulin, sondern auch ein Proteinhormon
namens Amylin/IAPP (daher der Name islet amyloid polypeptide). Im Prinzip verfügen Zellen
über standard operating procedures,
um mit einer solchen Situation fertigzuwerden: die unfolded protein response (UPR), die aus mehreren Stufen besteht. Zuerst
wird die Proteinsynthese generell heruntergefahren. Das reduziert im konkreten
Fall die Insulinproduktion. Zweitens wird jedoch die Synthese von
Chaperonproteinen gezielt verstärkt, um die korrekte Faltung neu synthetisierten
Proteins zu verbessern. Drittens werden fehlgefaltete Proteine aus dem ER ins
Zytosol exportiert und im Proteasom abgebaut. Wenn all das nichts nützt, wird schließlich
der programmierte Zelltod eingeleitet. Dazu kommt in diesem speziellen Fall noch,
dass Konglomerate von fehlgefaltetem IAPP eine Tendenz zu prionenartiger
Vermehrung zeigen und besonders toxisch wirken. Auf die Dauer reduziert dieser Prozess
die β-Zellkapazität im Pankreas und führt von Insulinresistenz zu
manifestem Diabetes mellitus Typ 2.
Auch im Pankreas tragen zusätzliche Mechanismen
zu dieser Entwicklung bei. Freie Fettsäuren (FFA, free fatty acids oder NEFA, non-esterified
fatty acids) induzieren in Pankreas-Inseln IL‑1β, IL‑6 und IL‑8. Die IL‑1β-Expression schaukelt
sich in einer positiven Rückkoppelung auf und behindert die Insulinsekretion.
Blockade dieser Rückkoppelungsschleife mit dem IL‑1-Rezeptorantagonisten
Anakinra verbesserte die β‑Zellfunktion und hatte niedrigere
HbA1c-Spiegel zur Folge.
Ohne es in allen Kausalitätsketten gänzlich zu
verstehen, nennen wir die Gesamtheit dieser Veränderungen Metabolisches
Syndrom. Insulinresistenz führt via Diabetes mellitus Typ 2 zu einem breiten
Fächer an Morbidität, wie Arteriosklerose, Herzinfarkt, Schlaganfall,
diabetische Nephro- und Retinopathie sowie Polyneuropathie.
Dieses Problem trifft uns nicht alle im
gleichen Ausmaß. In der genetischen Lotterie haben manche mehr, manche weniger
Energiespar-Allele gezogen. So berichtete eine Studie, dass ein Individuum mit
Energiesparallelen auf 104 von 194 untersuchten polymorphen Loci im
Durchschnitt 11 kg schwerer war als jemand mit nur 78 thrifty genes.
Was ist ein thrifty gene? Ein Beispiel: FTO-Gen und braunes Fettgewebe
Das Leben ist nicht gerecht: manche Menschen
können eine Menge essen, ohne zuzunehmen, die meisten können das nicht. Manche
Menschen haben nie zu kalt: entspannt sitzen sie kurzärmlig herum, während
andere frierend den Reißverschluss ihrer Jacke noch etwas höher zu ziehen
versuchen. Könnte es sein, dass manche Menschen einfach mehr Nahrung
"verheizen"?
Bis vor wenigen Jahren nahm man an, dass
braunes Fettgewebe nur bei Babys vorkommt. Fett ist gewöhnlich weiß; enthalten
spezialisierte Ansammlungen von Fettzellen allerdings einen hohen Anteil an
Mitochondrien, erscheinen diese beige oder braun. In den Mitochondrien verbrennen
diese Zellen Fett oder Glucose, um Wärme zu erzeugen. Normalerweise benützen
Mitochondrien die Energie eines Protonengradienten, um ATP zu synthetisieren,
wie in einer Mühle die Energie eines Baches verwendet wurde, um Korn zu mahlen. Würde
der Müller das Räderwerk vom eigentlichen Mühlstein entkoppeln, produzieren die
nun schneller ratternden Zahnräder nur mehr Wärme, keine Arbeit. In ähnlicher
Weise wird der Fluss von Protonen vom Antrieb der ATP-Synthase entkoppelt durch
die Expression von uncoupling protein 1 (UCP1 oder Thermogenin), indem dieses den Protonen ermöglicht, durch die innere
Mitochondrienmembran zurückzurieseln. Beige oder braune Fettzellen sind so in
der Lage, ihre Fettverbrennung von der ATP-Produktion abzukoppeln und
stattdessen zur Erzeugung von Wärme einzusetzen. Dieser Prozess wird durch ein
Zusammenspiel von sympathischer Aktivierung und Schilddrüsenhormon gesteuert. Aktivierung
β3-adrenerger Rezeptoren induziert das Enzym Iodothyronin-Deiodinase
Typ II, ein Selenoprotein, das Thyroxin (T4) zum aktiven Trijodthyronin (T3) umwandelt.
T3 aktiviert den bereits auf dem Promotor des UCP1-Gens sitzenden
Schilddrüsenhormonrezeptor.
Dann erkannte man mit Hilfe der 18F-Fluordesoxyglucose-PET-CT-Technik,
dass beiges oder braunes Fett auch bei Erwachsenen vorkommt, in kleinen
Ansammlungen im vorderen Hals und im Thorax. Anfangs fand man es nur bei
wenigen, sehr schlanken, Menschen und bei manchen Probanden, die man der Kälte
ausgesetzt hatte. Mit der Zeit wurde klar, dass Kurzzeit-Kälteexposition die
Aktivität braunen Fettgewebes steigert, Langzeit-Kälteexposition (Wochen) auch
das Volumen. Bei ausreichend rauer Kälteexposition kann braunes Fettgewebe in
praktisch jeder Person nachgewiesen werden. Doch schlanke Menschen haben im Allgemeinen
mehr braunes Fettgewebe.
Unabhängig davon schritt die Suche nach
Übergewicht-fördernden Genvarianten mit Hilfe von genome-wide association studies voran. In den langen Listen so
identifizierter Gene stand wiederholt ein Gen unklarer Funktion an der Spitze,
das wir nun fat mass and obesity
associated (FTO) nennen. Unklar blieb auch, über welchen Mechanismus
bestimmte Allele dieses Gens zur Fettleibigkeit beitragen. Dann wurde erkannt,
dass das kodierte Protein DNA-Service-Funktion hat, indem es Methylierungen von
Thymidin rückgängig macht. Trotzdem brachte das die Suche nach der Verbindung
des Gens mit dem Phänotyp "Übergewicht" nicht wirklich weiter.
Im Jahr 2015 wurde schließlich ein unerwartetes
Modell präsentiert: Das FTO-Gen trägt nicht durch sein kodiertes Protein zur Entstehung
von Übergewicht bei, sondern durch eine Bindungsstelle in Intron1 für einen
Transkriptionshemmer, der auf Gene weiter unten am Chromosom wirkt. An einer
bestimmten Stelle auf Chromosom 16, zwischen den FTO-Exons 1 und 2, hat ein SNP
(single nucleotide polymorphism rs1421085) zur Folge, dass manche von uns ein C (nennen wir es "Cnauser-C"),
andere ein T ("Thermo-T") tragen. Jede(r) von uns hat natürlich zwei Allele,
sodass unser Genotyp CC, CT oder TT sein kann. Der Einfachheit halber sehen wir
uns nur die CC und TT-Genotypen näher an (CT liegt irgendwo dazwischen).
1.
Sie sind
ein Thermo-TT-Typ? Da haben Sie Glück, denn Sie tun sich leichter, Fett zu
verbrennen. Dafür ist ein komplexer Mechanismus verantwortlich. Thermo-T ist
Teil einer Bindungsstelle für den Transkriptionshemmer ARID5B. ARID5B hemmt die
Transkription zweier Gene, die ein Stück weiter auf Chromosom 16 liegen, IRX3
und IRX5, die selbst wieder Transkriptionshemmer kodieren. In der
Differenzierung neuer Fettzellen fungieren IRX3 und IRX5 als Schalter zwischen
braun und weiß, als "Braun-Ausschalter". Thermo-T hemmt diese Hemmer,
sodass größere Mengen braunen Fettgewebes entstehen. Sind Sie Thermo-TT,
verheizen Sie mehr Fett für Wärme. Vielleicht sind Sie sogar der Typ, der
kurzärmlig in der Kälte sitzt! AAABER hätten Sie in der Altsteinzeit gelebt,
wären Sie weniger gut dran gewesen: da Sie dauernd mehr Kalorien verheizen,
wären Sie in einer Hungerperiode vielleicht unter den ersten gewesen, die den
Löffel abgeben.
2.
Sie sind
ein Cnauser-CC-Typ? Dann sind Sie ausgezeichnet ausgestattet für
altsteinzeitliche Lebensumstände. Cnauser-C verhindert jegliches Andocken des
Transkriptionshemmers ARID5B. IRX3 und IRX5 werden ungehindert stark exprimiert
und unterdrücken in neu entstehende Fettzellen das Bräunungsprogramm. Fast ausschließlich
weißes Speicherfettgewebe wird gebildet, sodass Sie in guten Zeiten reichlich Polster
aufbauen können. Bessere Aussichten, die nächste Hungersnot zu überleben!
So, nun verstehen wir ein thrifty gene. Doch es
gibt mehr als vierhundert davon, und jedes trägt nur einen kleinen Teil zur beobachteten
Gesamtvarianz im Body-Mass-Index bei.
Abhängig vom Satz an Allelen, den wir in der großen genetischen Lotterie von
unseren Eltern erhalten haben, sind wir im Umgang mit Nahrungsmittelenergie
eher knausrig oder eher hemdsärmelig, und wir können bisher nichts daran
ändern. Die beneidenswerten Zeitgenossen mit weniger Energiesparallelen bleiben
unter unseren Ernährungsbedingungen ohne besondere Anstrengungen schlank; die
Mehrheit muss sich bewusst bemühen, um das Gewicht zu halten.
Ein anderer übergewichtsfördernder Aspekt
des paläolithischen Programms: Zucker- und fettreiche Nahrungsstoffe (mmmh,
Schokolade!) lösen im ZNS ähnliche Belohnungsmuster aus wie Kokain und andere
Drogen. Bei der damaligen minimalen Verfügbarkeit hochkalorischer Nahrung war diese
verhaltenssteuernde Wirkung hilfreich; heute ist sie ein Problem. Nahrungsmittelhersteller
und -händler, die damit Gewinn machen, nützen diese Schwäche mit verführerischer
Werbung nach Kräften aus.
Der Beitrag unserer Mikrobiota
Ein zweiter Aspekt, in dem sich Individuen unterscheiden, ist ihre Ausstattung mit Darmbakterien, in ihrer Gesamtheit früher Darmflora, heute meist Mikrobiota genannt (ursprünglich ein Pluralwort, doch schließe ich mich dem allgemeinen Gebrauch als Einzahlwort an). Die Mikrobiota Übergewichtiger ist klar anders zusammengesetzt als jene schlanker Menschen. Weniger klar ist, was Ursache und was Wirkung darstellt. Der menschliche Darm wird durch eine komplexe Gemeinschaft von Bakterien und Archaea bewohnt, die sich aus insgesamt mehr als 1000 Spezies rekrutieren kann. Jede(r) von uns beherbergt mehr als 1013 solcher Zellen aus mindestens 160 Spezies. Ungefähr 60 Spezies finden sich in mehr als 90% aller Europäer. Ausgehend von Tierversuchen wurde mehrfach beschrieben, dass das quantitative Verhältnis zweier Phyla, der Firmicutes (z. B. Clostridium, Lactobacillus) und der Bacteroidetes (z. B. Bacteroides, Xylanibacter), mit der Ausbildung von Übergewicht korreliere: demnach dominierten Firmicutes in übergewichtigen, Bacteroidetes in schlanken Individuen. In Metaanalysen von Humanstudien zeigte sich diese Assoziation jedoch nicht konsistent.
Mehrere Faktoren beeinflussen die Zusammensetzung
der Mikrobiota. Den wichtigsten Faktor stellen die Ernährungsgewohnheiten eines
Menschen dar.
Die Hypothesen auf diesem Gebiet basieren auf Experimenten mit unter Laborbedingungen keimfrei aufgezogenen Mäusen. Diese keimfreien Mäuse können mit definierten mikrobiellen Spezies oder mit "Stuhltransplantaten" von Mäusen oder Menschen kolonisiert werden, um mögliche physiologische oder pathologische Auswirkungen zu überprüfen. Keimfrei aufgezogene Mäuse sind vor Übergewicht und Metabolischem Syndrom geschützt. Werden sie mit konventioneller Mikrobiota kolonisiert, steigt ihr Körperfettanteil jedoch merklich an. Keimfreie Mäuse, die mit Mikrobiota von adipösen Mäusen oder Menschen kolonisiert werden, die häufig z. B. das Archaeon Methanobrevibacter smithii enthalten, nehmen stärker zu als Mäuse, die Mikrobiota von schlanken Individuen erhalten. Wie ist das möglich?
Die Mikrobiota kann auf zumindest vier Arten den
Metabolismus beeinflussen:
1.
durch verstärkte Ausbeutung der in
der Nahrung enthaltenen Energie. Im Prinzip sind wir mit diesem Konzept bereits
vertraut: denken wir an wiederkäuende Rinder. Die Kuh selbst kann Gras
eigentlich ebensowenig verdauen wie wir, doch erhält sie Hilfe durch Bakterien
in ihrem dafür spezialisierten Verdauungstrakt. Unverdauliche
Kohlenhydratbestandteile des Grases, wie Zellulose und Xylan, werden in ihren
abgeteilten Mägen fermentiert, sodass ein großer Teil der darin enthaltenen
Energie für die Kuh verfügbar wird. Bei Mäusen oder Menschen ist dieser Prozess
weit weniger effizient, da die meisten Bakterien im Kolon sitzen und damit
hinter dem Dünndarm, wo die meisten Energieträger absorbiert werden. Abhängig
von der individuellen Mikrobiota können jedoch auch im Kolon durch Fermentation
unverdaulicher Kohlenhydrate kurzkettige Fettsäuren (Azetat, Propionat,
Butyrat) gebildet werden, die dort noch als Energieträger aufgenommen werden
können. So kann, abhängig von der Mikrobiota, mehr oder weniger Energie aus der
Nahrung extrahiert werden.
2.
durch direktes Drehen an
physiologischen Stellschrauben des Gastorganismus. Neben ihrer Funktion als Energieträger
aktivieren bestimmte kurzkettige Fettsäuren die G-Protein-gekoppelten
Rezeptoren Gpr41 und Gpr43. Aktivierung von Gpr41 führt zu Veränderungen in der
Sekretion von PYY (weiter unten beschrieben), Aktivierung von Gpr43 zu
Veränderung in der Sekretion von glucagon-like
peptide durch L-Zellen im Darmepithel. Ein zweites Beispiel:
Darmepithelzellen von keimfreien Mäusen produzieren beträchtliche Mengen von fasting-induced adipose factor (Fiaf
oder ANGPTL4). Fiaf, ein Glycoprotein, hemmt die Lipoproteinlipase (LPL),
welche die Aufnahme von Fettsäuren aus Chylomikronen und VLDL in Adipozyten
vermittelt. Kolonisierung keimfreier Mäuse mit konventioneller Mikrobiota
unterdrückt die Fiaf-Produktion in Darmepithelzellen; die Mäuse werden fetter.
Werden keimfreie Mäuse jedoch spezifisch mit Lactobacillus paracasei kolonisiert, bleibt der Fiaf-Spiegel höher.
3.
durch proinflammatorische Wirkung
auf den Gastorganismus. Das Zusammenspiel von individuellen Ernährungs-,
Mikrobiota- und genetisch bedingten Wirtsfaktoren kann die Durchlässigkeit des
Darmepithels steigern. In dieser Situation der erhöhten Permeabilität können
Spuren von Lipopolysacchariden und DNA der Mikrobiota über die Pfortader die
Leber erreichen und dort Toll-like
receptors 4 und 9 aktivieren. Dieser pro-inflammatorische
Effekt kann die Entwicklung von Insulinresistenz und nichtalkoholischer
Fettleber fördern.
4. durch Beeinflussung von im ZNS generierten Nahrungspräferenzen über von mikrobiellen Molekülen ausgelöste vagale Afferenzen.
Inwieweit Mäusebefunde auch auf die
Situation des Menschen zutreffen, muss weiter geklärt werden. Lange bekannte
Effekte, wie die positive Wirkung von ballaststoffreicher Ernährung, beginnen,
über die Microbiota verständlich zu werden. So förderte eine definierte
faserreiche Ernährung bei Diabetes
mellitus Typ 2-Patienten die Ausbreitung von Mikrobiota-Spezies, die zu einer
verstärkten glucagon-like peptide 1-Produktion
und zu einer Verminderung des HbA1c-Werts führten. Wenn wir
versuchen, unsere Tendenz zur Entwicklung von Übergewicht zu verstehen,
müssen wir in unsere Überlegungen jedenfalls nicht nur unsere genetische
Ausstattung, sondern auch unsere Mikrobiota und die Mikrobiota-steuernden
Eigenschaften unserer Ernährung einbeziehen.
Obwohl die Zeit für evolutionäre Veränderungen
seit dem Auszug aus Afrika knapp war, hat sich der veränderte Selektionsdruck
doch hier und da ausgewirkt. Es gibt Hinweise darauf, dass Gesellschaften,
in denen die bis dahin vorherrschende Lebensmittelknappheit erstmals
in ihr Gegenteil umschlug, massive Morbiditätskrisen durchmachten. Solche
Phasen gab es in Europa z. B. während der Römerzeit, als gut organisierte
Landwirtschaft und Transportsysteme die durchschnittliche Versorgungssituation
massiv verbesserten. Wahrscheinlich starben damals viele Menschen jung
an Diabetes mellitus und damit zusammenhängenden Gesundheitsproblemen.
In diesen Gesellschaften begann damals erstmals eine Selektion gegen die thrifty genes. In Populationen, die dem kalorischen Überfluss heute
erstmals ausgesetzt sind, finden wir zurzeit die stärksten Probleme
bezüglich Übergewicht und Diabetes mellitus Typ 2.
Das Rezept für Gewichtsabnahme ist
einfach: nimm weniger Kalorien auf als du verbrauchst. Allerdings
sind wir darauf konstruiert, den Zustand einer negativen Energiebilanz
mit allen Mitteln zu vermeiden. Rasch werden Energiesparprogramme hochgefahren,
die uns passiv und grantig (hochdeutsch: griesgrämig) machen, frieren
lassen, unsere Gedanken ständig ums Essen kreisen lassen und uns bereit
machen, uns selbst hemmungslos zu belügen. Darum gelingt es nur wenigen,
lediglich durch Zurückhaltung beim Essen abzunehmen. Dauerhafte Gewichtsabnahme
gelingt in der Regel nur durch weitreichende Umstellung der Alltagsgewohnheiten
in Bezug auf Ernährung, sozialen/Trinkgewohnheiten und Bewegung. Regelmäßiges
Ausdauertraining wirkt dem Energiesparmodus entgegen.
Gewichtsabnahme steht weit oben auf
der Prioritätenliste unserer Gesellschaft, wenn auch mehr aus ästhetischen
als aus Gesundheitsgründen. Leider wird den Leuten viel zu häufig mit
schlechtem Rat und fragwürdigen Produkten Geld aus der Tasche gezogen.
Im Folgenden daher eine (gratis!) evidenzbasierte Liste von Empfehlungen,
die dabei helfen können, dauerhaft abzunehmen. Viele dieser Aspekte
werden später noch näher erklärt.
Vor allem: das Körpergewicht ist ein Resultat der Energiebilanz. Abnehmen bedeutet, weniger
Kalorien zuzuführen als zu verbrauchen. Keine Diät, kein Trick führt
an diesem Grundsatz vorbei.
Die Energiezufuhr-Seite:
1.
Am Anfang steht der Entschluss, über
eine lange Zeit etwas weniger zu essen, als man braucht. Intensive Kurzzeit-Diäten
sind nicht nachhaltig: man quält sich einige Wochen, sehnt den Tag herbei,
an dem es endlich vorbei ist und legt damit schon den Grundstein für
den Jo-Jo-Effekt.
2.
Ein Langzeit-Marschplan mit realistischen
Zielen hilft, diese zu erreichen: 0,5 kg pro Monat ist machbar und
fördert die notwendige Umstellung der Lebensgewohnheiten.
3.
Eine tägliche Hungerphase ist gut!
Solange man nach einer Mahlzeit Kalorien aufnimmt, dominiert Insulin,
das die Umwandlung eines Teils dieser Kalorien in Fett steuert. Sobald
sich dieser Prozess umkehrt, dominieren Sympathikusaktivierung und Glucagon;
man wird wieder hungrig. Hunger ist das Signal an den Altsteinzeitmenschen,
wieder auf Nahrungssuche zu gehen: erst die Bewegung, dann die nächste
Mahlzeit. Solange man hungrig ist, verbrennt man Fett. Die häufigen
"Abnehmen, ohne zu hungern!"-Slogans kommen von Leuten, die
daran verdienen wollen.
4.
Frühstückskalorien werden eher verbraucht,
Abendessenskalorien eher gespeichert: Tageskalorien daher zugunsten des
Frühstücks verteilen! Es ist nicht notwendig, eine
Mahlzeit auszulassen; will man das trotzdem, verzichtet man am besten
auf das Abendessen. Um zu
verhindern, dass Insulin – das die Lipolyse hemmt – zu häufig aktiv ist, sollte
man zwischen den Mahlzeiten nicht naschen und über Nacht nichts essen (z.B.
8/16-Intervallfasten).
5.
Als Getränke empfehlen sich Wasser
und Tee. Auf Säfte sollte man verzichten, den Alkoholkonsum zurückschrauben.
Viele Kalorien nehmen wir in flüssiger Form zu uns. Erfrischungsgetränke
enthalten große Mengen Zucker: Fructose, Glucose, Saccharose.
Alkohol (7 kcal/g) verstoffwechseln wir zu Acetyl-CoA und Reduktionsäquivalenten,
also zu den Ausgangsprodukten der Fettsäuresynthese. Immer, wenn wir
Alkohol trinken, synthetisieren wir Fett, statt es zu verbrennen.
6.
Selbst kochen! Dann weiß man, was man
isst.
7.
Kleinere Portionen servieren, kleinere
Teller verwenden wegen des Portionsgrößeneffekts.
8.
Primäre Lebensmittel helfen bei der
Kalorienreduktion: Obst, Gemüse, Hülsenfrüchte, Salate, mageres Fleisch,
Fisch. Industriell verarbeitete Lebensmittel haben in der Regel eine
höhere Energiedichte, d. h., sie enthalten zu viel Fett und Zucker.
Außerdem enthalten sie häufig zu viel Salz, Trans-Fette und andere Stoffe
zweifelhafter Nützlichkeit.
9.
Eiweißreiche Mahlzeiten machen es leichter,
die Gesamtkalorienzufuhr zu vermindern. Verglichen mit isokalorischen
Alternativen zögert eine eiweißreiche Mahlzeit über Ausschüttung von
PYY die Rückkehr des Hungergefühls hinaus. Zu den proteinreichen Nahrungsmitteln
gehören Eier, Fisch und mageres Fleisch; Vegetarier können auf Hülsenfrüchte, speziell Sojabohnen/Tofu,
Kerne (Kürbis, Sonnenblume) und Nüsse zurückgreifen.
10.
Mahlzeiten mit niedrigem glykämischen
Index unterstützen Abnehmbemühungen. Dieser Punkt überlappt mit den
vorigen beiden, denn primäre und proteinreiche Lebensmittel tendieren
zu einem niedrigeren glykämischen Index. Konkret bedeutet das Zurückhaltung
bei zuckerhaltigen Lebensmitteln wie Süßigkeiten, Schokolade, Konditoreiwaren.
Den glykämischen Index des Frühstücks kann man herabsetzen, indem man
Weißbrot und industrielle Frühstücksflocken durch Vollkornmüsli mit
Obst und Nüssen ersetzt.
Die Energieverbrauchs-Seite:
11.
Mehr tägliche Routinebewegung erhöht
den Energieverbrauch: zur Arbeit gehen oder Rad fahren statt mit Bus
oder Auto; die Stiege benützen statt den Lift.
12.
Bewegung verbraucht nicht nur unmittelbar
Energie, sie erhöht den Grundumsatz auch für viele Stunden danach. Empfohlen
sind 150 min pro Woche, intensive Minuten zählen doppelt und haben einen
stärkeren Nachbrenneffekt. Die
Form ist sekundär: flott gehen, joggen, Rad fahren, schwimmen, langlaufen,
tanzen
13. Hält man Innentemperatur und Kleidung eher kühl, verheizt der Körper selbst mehr Energie durch adaptive Thermogenese in Muskel und braunem Fettgewebe.
Um Gewicht abzubauen, ist (zu) wenig zu essen wichtiger als viel Bewegung: You can't outrun a bad diet. Sorgfältige Untersuchungen des Energiehaushalts (mittels der Gold-Standard doubly labeled water-Methode) der als Jäger und Sammler lebenden Hadza-Population in Tanzania brachten eine unerwartete Erkenntnis: Diese Menschen, die sich täglich für die Nahrungssuche sehr viel bewegen, verbrauchen pro Tag nur unwesentlich mehr Kalorien als wir Schreibtischmenschen. Werden viele Kalorien für Bewegung benötigt, findet der Körper offensichtlich anderswo Einsparungsmöglichkeiten. Wir kennen das von den positiven Gesundheitseffekten von Sport: wenn wir regelmäßig trainieren, sinkt unsere mittlere Herzfrequenz, sinkt unser mittlerer Blutdruck; das spart Energie.
Unsere
intuitive Vorstellung von Gewichtsabnahme sieht etwa so aus: Wir essen
eine Weile weniger, bis wir unser Zielgewicht erreicht haben, und dann
essen wir wieder "normal", das heißt, ähnlich viel wie vorher.
Diese Vorstellung ist leider aus zwei Gründen falsch:
1.
Der Energieverbrauch
(Kalorienbedarf) unseres Körpers ist eine Funktion seiner Masse. Wer
das nicht glaubt, schleppe einmal für einige Tage einen 10 kg-Rucksack
herum. Wenn wir also erfolgreich abnehmen, hat unser Körper am Zielgewicht
einen geringeren Kalorienbedarf als vorher. Beispiel: Eine 22-Jährige,
165 cm, empfindet ihre 70 kg (BMI 25,7) als zu viel und nimmt mit viel
Mühe und Rückschlägen, aber letztlich erfolgreich, 10 kg ab. Ihr 60
kg-Körper (BMI 22) hat, nach Standardformeln berechnet, einen Kalorienbedarf
von 2170 kcal pro Tag, während ihr 70 kg-Körper 2340 kcal zu sich nehmen
konnte, ohne weiter zuzunehmen. Sie muss also zumindest um die Differenz
von 170 kcal weniger essen als vorher, um nicht wieder zuzunehmen.
2.
Die tatsächliche Differenz
ist allerdings noch höher: Unser Organismus "merkt" sich das
höhere Ausgangsgewicht und setzt alles daran, dieses wieder zu erreichen.
Ein einmal für längere Zeit erreichtes
Gewichtsmaximum wird als neuer Sollwert gespeichert, der vom Organismus mit
Zähnen und (Ver-) Dauen verteidigt wird. Bei Personen, die mehr als 10 %
ihres Körpergewichts abgenommen hatten und dieses Gewicht einigermaßen halten
konnten, fand man, dass ihr Organismus das neue Gewicht ein volles Jahr später
immer noch als Defizitzustand interpretierte: Schilddrüsenhormon sowie die
Sattheitshormone Leptin und PYY waren erniedrigt, das Hungerhormon Ghrelin
erhöht und die Testpersonen gaben verstärktes subjektives Hungergefühl an.
Sorgfältige Messungen ergaben, dass der Gesamtenergieverbrauch solcher Personen
geringer ist, als der von gleich schweren Vergleichspersonen, die nicht
abgenommen haben. Das ist zum Teil darauf zurückzuführen, dass die Phase des
Übergewichts epigenetische Veränderungen im Fettgewebe zurücklässt, die auch
noch lange nach Gewichtsabnehme bestehen bleiben: Fettzellen von
"Post-Übergewichtigen" nehmen Energieträger rascher auf und bleiben
zögerlicher, die Lipolyse wieder hochzufahren, was den Yo-yo-Effekt fördert.
Ähnliche Mechanismen dürften dafür verantwortlich sein, dass auch die Muskeln
im Alltagsleben energieeffizienter arbeiten, da diese sich tendenziell in Richtung sparsame Typ I-Fasern (slow-twitch, myosin heavy chain I)
und weg von glykolytischen Typ II-Fasern (fast twitch, myosin heavy chain IIa und IIx) entwickeln.
Auf das Beispiel der jungen Frau umgelegt:
hätte sie nie ein höheres Gewicht gehabt, würde ihr 60 kg-Körper die
berechneten 2170 kcal pro Tag verbrauchen. Ihr "post-70 kg"
60 kg-Körper befindet sich ein Jahr später allerdings immer noch im
Energiesparmodus und verbraucht weniger. Diese zusätzliche Reduktion
wurde bisher nur in wenigen Probanden quantifiziert und variierte stark,
sodass es nicht möglich ist, in unserem Beispiel eine exakte Zahl anzugeben,
doch liegt sie vermutlich zwischen 100 und 400 kcal.
Der neue Gleichgewichts-Energieverbrauch der jungen Frau liegt damit irgendwo zwischen 1770 und 2070 kcal pro Tag. Mit anderen Worten: Will sie ihr Gewicht halten, muss sie bei verstärktem Hungergefühl jeden Tag um 270-570 kcal oder etwa eine halbe Mahlzeit weniger essen, als ihrem Gefühl von "normal" entspricht. Darum also ist es so schwierig, nach einer Gewichtsabnahme das Zielgewicht zu halten. Eine wesentliche Schlussfolgerung: Prävention von Übergewicht ist um vieles leichter als nachträgliche Korrektur. Prävention von Übergewicht, sowie von Rauchen, sind unspektakulär und unbedankt (das "Präventionsparadox"), gehören aber zu den wirksamsten ärztlichen Aufgaben.
"Entschlackung"?
Als "Schlacke" bezeichnet
man Gesteinsumformungen, die bei der Verhüttung von Erzen bei der
Metallgewinnung übrigbleiben. In unserem Organismus gibt es nichts Analoges.
Wenn uns eine Entschlackung angepriesen wird, bedeutet das in der Regel, dass
wir eine Dienstleisung oder ein Produkt kaufen sollen, meist in Verbindung mit
Fasten. Fasten selbst ist im Übrigen nicht teuer und kann in moderater Form,
z.B. als Intervallfasten, durchaus sinnvoll sein.
Beim Fasten werden die dauernd
benötigten Zucker, Fette und Aminosäuren nicht mehr über den Darm aufgenommen.
Über die notwendige Umstellung der Energiebereitstellung machen wir uns weiter unten
– Abschnitt Leber – Gedanken. Doch wie werden unsere Zellen mit dem Mangel an
Aminosäuren fertig? Zelluläre Proteine haben unterschiedliche Halbwertszeiten,
und viele von ihnen müssen wir dauernd nachsynthetisieren, um die Zellen
funktionsfähig zu erhalten.
Unsere erste ernste Fastenzeit haben
wir alle bereits hinter uns: jene als Neugeborene. Mit dem Durchtrennen der
Nabelschnur ging uns nicht nur der Sauerstoff aus, was uns zu schmerzhaften
ersten Atemzügen zwang. Vorbei war es auch mit der anstrengungslosen Anlieferung
von Nährstoffen. Leider dauerte es einige Tage, bis bei unserer Mama die
Muttermilch einschoss. Für uns hieß das: darben und schrumpfen! Wie kamen wir
über die Runden? Das Geheimnis heißt Autophagie. Um weiter Proteine
synthetisieren zu können, schmolzen unsere Zellen große Schlucke von
zytosolischer Proteinsuppe ein und verdauten diese zu Aminosäuren. Technisch
funktioniert das so, dass Doppelmembranballons um Schlucke von Zytosol gebildet
werden, die dann mit Lysosomen fusionieren, sodass die Proteine zu Aminosäuren
recycelt werdern. Es war eine Notmaßnahme, aber es hielt unsere Zellen am Leben
und damit unseren Organismus. Erst nach ein oder zwei Wochen erreichten wir
wieder unser Geburtsgewicht, was unsere Mütter mit Stolz vermerkten.
Auch wenn wir als Erwachsene fasten,
setzen wir diese nicht-selektive Autophagie in Gang. Werden wichtige
Aminosäuren in der Zelle knapp, bremst das den mTOR (mechanistic target of rapamycin)-Komplex 1. Das aktiviert die
Autophagie und bremst wiederum viele Zellfunktionen. In vielen Spezies,
wahrscheinlich auch beim Menschen, verlängert lebenslange Energieknappheit die
Lebenszeit. Ob und wenn, inwieweit das mit dem beschriebenen Mechanismus zu tun
hat, bleibt leider vorerst noch unklar.
Als Gegensatz zur nicht-selektiven
existiert auch die selektive Autophagie, die dazu dient, ins Zytosol
eingedrungene Bakterien oder nicht mehr gebrauchte Zellorganellen abzubauen.
Dieser Prozess wird nicht durch Fasten angestoßen, sondern durch das Auftreten
von partikulärem Material, das entsorgt gehört und mittels zytosolischen
Rezeptoren erkannt wird. Dies wäre am ehesten mit Entschlackung vergleichbar,
ist aber nicht von außen steuerbar.
Kurz: Wird Ihnen Entschlackung
angepriesen, reduzieren Sie lieber privat Ihre Nahrungszufuhr. Das ist
weit billiger.
Ähnlich, wie wir für einen Kalorienmangel
genetisch besser vorbereitet sind, als für einen Kalorienüberschuss,
sind wir für einen Salzmangel besser vorbereitet, als für Salzüberschuss.
Während wir mit dem Aldosteronsystem einen sehr effizienten Mechanismus
haben, Salz zu sparen, sind unsere Mechanismen zur Salzausscheidung
weniger effizient, weil sich dieses Problem in der Evolution des modernen
Homo sapiens nicht stellte. Der evolutionär
begründete Appetit nach Salz in Kombination mit dessen reichlicher Verfügbarkeit
führt heute zu Überkonsum. Mehr Salz führt bei ineffizienter Ausscheidung
aus osmotischen Gründen zu mehr Volumen, und tendenziell höheres Volumen
im geschlossenen System des extrazellulären Raums bedeutet tendenziell
höheren Blutdruck. Erst mit Hilfe des höheren Blutdrucks sind wir in
der Lage, mehr Salz auszuscheiden, da die renale Natriumausscheidung
eine Funktion der tubulären Flussrate ist (pressure
natriuresis). Unsere hohe Salzaufnahme begünstigt also die Entstehung
von Bluthochdruck. Die DASH-Ernährungsempfehlung
(Dietary Approaches to Stop Hypertension,
Link leider nur auf Englisch verfügbar,
mit englischen Maßen und Gewichten) zielt auf diesen Punkt ab und
stellt generell einen guten Ratgeber zur gesunden Ernährung dar.
4. UNTERERNÄHRUNG
Unterernährung durch Essstörungen: Anorexia und Bulimia nervosa
Ab Crack, Bikini Bridge, Hip Cleavage, Ribcage Bragging, Sixpack, Thigh
Brow, Thigh Gap, Toblerone Tunnel: Wer weiß, was alle diese Begriffe bedeuten, ist vermutlich einmal
falsch in Richtung Sofort-Gram (Insta-Gram) abgebogen. TokTok, tippen sich Andere
ungläubig ans Hirn. Soziale Medien verstärken, vor allem bei jungen Menschen,
die ihre Mitte noch nicht gefunden haben, den Druck, vorgegaukelten Phantasie-Idealen zu entsprechen. Essstörungen betreffen am
häufigsten junge Frauen, weniger häufig aber auch andere Altersgruppen und
Männer. Typisch dabei ist eine Körperschemastörung – eine veränderte
Wahrnehmung des eigenen Körpers. Während die Umwelt auf die Knochen abgemagerte
Untergewichtige sieht, erleben sich die Betroffenen als "zu dick".
Hier wird einmal mehr klar, dass unser ZNS unsere individuelle "Welt"
konstruiert und mit Hilfe dieses Modells unsere Sinneswahrnehmungen
interpretiert. Solange die Weltmodelle zwischen Menschen, die miteinander
interagieren, weitgehend überlappen, fällt das nicht auf. Umso mehr macht es
uns ratlos, wenn die Divergenz zu groß wird, wie im Beispiel der Anorexia nervosa.
Körperliche Folgen sind gravierend bis
lebensbedrohlich. Der Stoffwechsel ist in den extremen Energiesparmodus
heruntergefahren. Die Betroffenen frieren ständig, einerseits durch geringe
Wärmeproduktion, andererseits durch das fehlende isolierende
Unterhautfettgewebe. Blutdruck und Herzfrequenz sind abgesenkt, häufig mit
QT-Verlängerung und entsprechendem Risiko für Herzrhythmusstörungen.
Hypokaliämie, ausgelöst durch Laxantien-, Diuretikaabusus und Alkalose durch
Erbrechen, steigert das Risiko für Rhythmusstörungen und – gemeinsam mit
Volumenmangel – Niereninsuffizienz. Mangels Material produziert das Knochenmark
zuwenig Blutkörperchen: Anämie, Leukopenie, Thrombopenie. Bei den Blutwerten
finden sich auch Hypoglykämie und weitere Elektrolytstörungen, die wiederum
Herzrhythmusstörungen fördern. Osteoporose durch Proteinmangel erhöht das
Frakturrisiko. Schilddrüsenhormon und Geschlechtshormone sind vermindert, mit
Amenorrhoe bei Frauen, Potenzverlust bei Männern. Haare werden dünn, brüchig,
fallen aus oder werden zu Lanugohaaren; Bulimie ruiniert die Zähne durch die Säureexposition.
Therapeutisch muss am "Weltmodell", inklusive der
Körperschemastörung, gearbeitet werden.
Unterernährung durch Ressourcenknappheit
Trotz des Überflusses in einem Teil
der Welt (etwa
400 Millionen leiden an Diabetes mellitus Typ 2, mehr als 1 Milliarde an Adipositas) ist Hunger nach wie vor eines der größten medizinischen Probleme.
Etwa 800 Millionen Menschen sind unterernährt. Laut Welternährungsorganisation
FAO verhungert alle fünf Sekunden ein Kind.
Ein großer Teil des Hungers ist menschengemacht.
Dies gilt z. B. für den Klimawandel, doch auch für die Wirtschaftsmechanismen,
mit deren Hilfe wir Nahrungsmittel produzieren und verteilen.
[Zwei Beispiele:
1. Um planen zu können, haben sowohl
der französische Weizenbauer als auch der österreichische Großbäcker Interesse
daran, den Weizenpreis in einem halben Jahr zu fixieren. Sie könnten
sich also zu einer Lieferung zu einem bestimmten Termin zu einem bestimmten
Preis einigen. Einen solchen Vertrag nennen wir ein Warentermingeschäft.
Tatsächlich haben weder Bauer noch
Bäcker Zeit, einen Partner zu suchen. Es benötigt also Vermittler. In
der Praxis sieht der Ablauf etwa so aus: Der Landwirt zahlt dem Vermittler
einen bestimmten Betrag und erwirbt damit das Recht, seinen Weizen über
den Vermittler bis zu einem bestimmten Termin zu einem bestimmten Preis
zu verkaufen. Wir nennen das eine Put-Option. Auch der Bäcker zahlt
dem Vermittler einen gewissen Betrag und erwirbt damit das Recht, Weizen
bis zu einem bestimmten Termin zu einem bestimmten Preis zu kaufen.
Wir nennen das eine Call-Option. Eine Option ist also ein Geschäft,
bei dem ein Recht verkauft wird. Jener, der zahlt, erwirbt das Recht;
jener, der Geld bekommt, verpflichtet sich, die Gegenseite
des zugrundeliegenden Geschäfts zu übernehmen. Der für die Option bezahlte
Preis ist eine Art Versicherungsprämie, um den Weizen später zu einem
bestimmten Preis verkaufen bzw. kaufen zu können. Wie der Name Option
impliziert, muss das Recht nicht ausgeübt werden: ist der Marktpreis
zur Zeit der Ernte höher als der im Termingeschäft vereinbarte Ausübungspreis,
kann der Landwirt an den Meistbietenden verkaufen. Nicht-ausgeübte Optionen
laufen wertlos aus, die Prämie bleibt dem Verkäufer der Option. Die
Rechte und Pflichten könne auch weiterverkauft werden, sodass eine Börse
für diese Optionen entsteht. Hedge Fonds, Banken, Versicherungen, die
nicht das geringste Interesse am Weizen haben, erwerben und verkaufen
solche Optionen, wenn sie sich davon Gewinn erwarten. Ein zweiter Typ
des börsegehandelten Warentermingeschäfts, bei dem sich --im Gegensatz
zur halbseitig verpflichtenden Option-- beide Seiten zur Durchführung
des Geschäfts verpflichten, sind die sogenannten Futures.
Die meisten Teilnehmer an der Warenterminbörse
sind weder Produzent noch Endabnehmer. Sie haben lediglich ein Interesse
an einer möglichst großen Gewinnspanne, und werden es ausnützen, wenn
unvorhergesehene Ereignisse es erlauben, diese Spanne zu vergrößern.
Sperren zum Beispiel die russischen Behörden die Weizenexporte,
da die Trockenheit eine schlechte Ernte erwarten lässt, erfahren die
gut vernetzten Teilnehmer an der Warenterminbörse das etwas früher als
der österreichische Bäcker und die anderen Abnehmer. Die relativ überschaubare
Zahl an Marktteilnehmern aus der Finanzindustrie braucht sich gar nicht
explizit abzusprechen, allen ist klar, welches Verhalten den Gewinn
vergrößert: sie werden von diesem Augenblick an sehr zurückhaltend sein,
sich zu Lieferungen zu verpflichten. Wenn ein Verbraucher trotzdem eine
Lieferung fixieren will, muss er eben viel dafür zahlen. Das bedeutet,
der Ausübungspreis für neue Weizentermingeschäfte steigt, und zusätzlich
werden die Call-Optionen teurer. Während die Weizenverknappung aus Sicht
der Nahrungsmittelversorgung ausschließlich negativ ist, ermöglicht
sie manchen Teilnehmern am Warenterminmarkt große Gewinne. Der erwartete
Ernteausfall in Russland hätte den Weizenmarktpreis bereits angehoben,
doch der Warenterminhandel verstärkt diesen Preisanstieg nun noch zusätzlich.
Der österreichische Großbäcker ist zunächst durch seine Call-Option preislich
abgesichert, doch für das nächste Termingeschäft wird er schließlich
zähneknirschend mehr zahlen. Wir Konsumenten ärgern uns, dass das Brot
schon wieder teurer wird. Doch für die weltweit 1,2 Milliarden Menschen,
die mit weniger als einem Euro pro Tag überleben müssen, ist der resultierende
Preisanstieg existenzbedrohend.
Gibt es eine Lösung? Wahrscheinlich
würde es helfen, den Kreis der Teilnehmer an Nahrungsmitteltermingeschäften
einzuschränken. Gesetzliche Regelungen könnten direkt mit den Nahrungsmitteln
befassten Marktteilnehmern, wie Produzenten, Abnehmern und, so weit
als notwendig, Vermittlern, Termingeschäfte ermöglichen, vom physischen
Produkt losgelöste Spekulation jedoch verhindern. Solche gesetzlichen
Regelungen wurden bisher nicht einmal versucht und wären wohl nur gegen
starkes Lobbying der Finanzwirtschaft durchsetzbar.
2. Subventionen für landwirtschaftliche Exporte in reichen Staaten: Europa subventioniert seine Landwirtschaft aus öffentlichen Mitteln. Das ist notwendig, um die lokale Lebensmittelerzeugung zu stabilisieren und abzusichern, damit Europa nicht zu abhängig von Importen wird. Importabhängigkeit ist dann gefährlich, wenn internationale Krisen oder Lieferkettenprobleme die Versorgung von außen unterbrechen. Die landwirschaftlich geprägte Landschaft ist zudem Teil der europäischen kulturellen Identität. Nebenwirkung der Subventionierung ist jedoch in guten Zeiten ein Überschuss an landwirtschaftlichen Produkten, der zu Preisen an z. B. Länder in Afrika geliefert wird, welche die lokalen Preise unterbieten: Der durchschnittliche afrikanische Landwirt kann von vornherein weit weniger effizient produzieren und wird überdies nicht subventioniert. Wenn er aber seine Produktion durch die Konkurrenz billiger Importe nicht verkaufen kann, bleibt ihm nichts anderes übrig, als diese zurückzufahren oder einzustellen. Fallen die Importe einmal aus oder verteuern sie sich, kann die lokale Produktion jedoch nicht rasch wieder hochgefahren werden.
3. Ein weiterer Faktor, der Nahrungsmittel
tendenziell verteuert, ist die Herstellung von Biotreibstoff. E5 ist
die Bezeichnung für Benzin, das 5% Ethanol enthält, das durch Vergärung
von Nahrungsmitteln wie Weizen, Mais und Zuckerrüben gewonnen wird.
Zwar ist es möglich, Ethanol aus z. B. Zuckerrohr zu gewinnen, das in
Sumpfgebieten wächst und weniger Flächenkonkurrenz darstellt, doch ist
der weltweite Nahrungsmittelanteil an der Kraftstoffethanolerzeugung
heute de facto hoch.
Hat all das überhaupt mit Medizin zu
tun? Wenn Menschen unnötig daran sterben: ja!]
Als eine Folge ihrer Armut müssen viele
Menschen mit einer extrem eingeschränkten Nahrungspalette auskommen,
im Wesentlichen mit Mais oder Reis. Während die täglich verfügbare Menge
für den rein kalorischen Bedarf noch ausreichen mag, fehlen bei einer
solchen Ernährung essentielle Bestandteile, sodass Krankheiten auf die
Dauer unausweichlich und der Tod nur zu oft die Folge sind.
Mit einer regelmäßigen Versorgung mit
Kohlenhydraten ist unser Organismus in der Lage, Energie zu produzieren
sowie andere notwendige Kohlenhydrate und viele Lipide durch Umbau
herzustellen. Das Problem beginnt bei den Proteinen. Von den zwanzig
Aminosäuren können Erwachsene acht nicht selbst synthetisieren –Kinder noch
mehr--, sodass diese mit der Nahrung aufgenommen werden müssen. Mehrere von
diesen sind in Getreide, Reis oder Mais in nicht ausreichender Menge vorhanden.
Ein Protein ist eine Kette von Aminosäuren: es liegt im Wesen einer Kette, dass
ein einziges fehlendes Kettenglied die gesamte Kette unbrauchbar macht. Können
wir ausgehend von Getreideprotein Muskeln aufbauen? Unser mengenmäßig
wichtigstes Muskelprotein, myosin heavy chain-1, enthält in 1939 Aminosäuren
208 Lysine (10,7%). Ein Hauptprotein in Weizen, α -Gliadin, enthält in 296
Aminosäuren magere 2 Lysine (0,6%). Bei einseitiger Getreideernährung, und das
inkludiert Reis, werden wir also nicht viel Muskel aufbauen können. Dasselbe
gilt für Mais, in dem wenig Lysin und Tryptophan enthalten sind. Die
Armennahrung vieler Entwicklungsländer führt damit besonders für Kinder oft zu
einer Mangelernährung.
Kwashiorkor
Sobald eine Aminosäure limitierend
wird, kann der Organismus nicht mehr genügend Proteinmoleküle herstellen.
Von außen sichtbar wird das zuerst für jene Proteine, die wir in hohen
Kopienzahlen benötigen: Muskelproteine und Albumin. Albumin benötigen
wir, um den kolloidosmotischen Druck zu erhalten; ein Mangel führt zu
typischen Fußödemen und Aszites-bedingtem Hungerbauch. In einer auf
den ersten Blick paradox erscheinenden Reaktion ist die Leber durch
Fetteinlagerung vergrößert, da die Lipide durch Apoproteinmangel nicht
als VLDL in die Peripherie verbracht werden können. Mangelhaft pigmentiertes,
schütteres Haar und Dermatitis sind weitere typische Zeichen. Betroffene
Kinder und Erwachsene sind apathisch und ein leichtes Opfer für Infektionen,
da eine effektive Immunantwort auf die Produktion von Antikörpern und
Zellen angewiesen wäre. Der Begriff Kwashiorkor kommt aus der Ghanaischen
Ga-Sprache und bedeutet "die Krankheit, die ein Kind bekommt, wenn
ein neues Kind geboren wird". Durch das Abstillen des älteren Kindes
wird die ausgewogene Aminosäurezusammensetzung der Muttermilch durch
die mangelhafte der pflanzlichen Ernährung ersetzt. Selbstverständlich
wird die Unterernährung in diesen Situationen durch weitere, zusätzliche
Faktoren verstärkt: der Armennahrung fehlt verwertbares Eisen, Vitamin
B12 (siehe unten), Niacin und sie ist häufig durch Aflatoxin kontaminiert.
5. ZENTRALE APPETIT-REGULATION
Der Hypothalamus ist der Haupt-Schauplatz für die Appetitregulation, und ein wesentliches Schaltzentrum ist der Nucleus arcuatus. Hier treffen Versorgungs-Informationen aus der Peripherie ein, z. T. über neurale Afferenzen, z. T. über Proteinhormone wie Leptin und Ghrelin.
Portionsgrößeneffekt
Je größer die Portion ist, die vor
uns steht, desto mehr essen wir. Gibt man der Durchschnittsperson einen
Teller mit mehr, als sie essen kann, isst sie eine gewisse Menge davon.
Gibt man derselben Person einen größeren Teller mit einer noch größeren
Portion, isst sie mehr als das erste Mal. Wir sind weit davon entfernt,
die Mechanismen solcher zentralen Steuerungen zu verstehen, doch handelt
es sich beim Portionsgrößeneffekt wahrscheinlich um einen Teil des Programms
zur Anlage von Fettdepots in Zeiten des Überflusses.
Praktische Bedeutung: Abnehmen/Gewicht halten fällt leichter, wenn man kleinere Portionen/Teller auftischt.
Von Badezuber-artigen Popcornbehältern wird abgeraten.
Leptin
Leptin (von griech. leptos=dünn) füttert das Zentrale Nervensystem
mit Informationen über die vorherrschende Versorgungslage. Das ZNS reagiert darauf mit sinnvollen
Modifikationen des Essverhaltens, der allgemeinen Aktivität, des Stoffwechsels
inklusive des Knochenstoffwechsels, der Reproduktionsfunktionen etc. (Anorexia nervosa kombiniert z. B.
niedrige Leptinspiegel mit Amenorrhoe).
Das Signalprotein Leptin wird fast ausschließlich durch Adipozyten sezerniert (wir sprechen daher von einem Adipokin). Sein Langzeit-Plasmaspiegel ist proportional zum Fettgewebs-Volumen des Individuums. Um dieses Niveau gibt es eine von den täglichen Mahlzeiten abhängige Oszillation mit in der Regel einem Tief vor dem Frühstück und einem Hoch am späten Abend. Längere Tagesphasen ohne Nahrungsaufnahme ("Postresorptionsphasen", z. B. in der zweiten Hälfte der Nacht) sind mit einem Abfall des Leptins verbunden: Geht das Leber-Glykogen zurück, führt der damit verbundene Plasmaglucose- und Insulin-Abfall zu einer Reduktion des Leptinspiegels. Das ZNS reagiert darauf mit Hungergefühl und einer Aktivierung der "Stress-Achse" CRH-ACTH-Cortisol. Erhöhtes Cortisol in Verbindung mit niedrigem Insulin führt zu Gluconeogenese, sodass der Blutzuckerspiegel stabilisiert wird. Dauert der Zeitraum ohne Nahrungsaufnahme länger als einen Tag ("Hungerphase" oder Fasten, obwohl das Hungergefühl dann wieder verschwindet), ist das Leberglykogen aufgebraucht, der Leptinspiegel halbiert sich und der Körper schaltet in einen Sparmodus, den wir später besprechen, wenn wir uns mit der Leber befassen.
Leptin und
andere Signalmoleküle beeinflussen Neuronen im Nucleus arcuatus im Hypothalamus, neben dem Boden des 3. Ventrikels.
Das ist möglich, da der Nucleus arcuatus der Eminentia mediana benachbart ist, die
zu den zirkumventrikulären Organen gehört und außerhalb der Blut-Hirn-Schranke
liegt. Erniedrigte Leptinwerte führen zu einem starken Hungergefühl, normale Leptinspiegel
zu Zufriedenheit ("Sattheit") mit erhöhter Bereitschaft zu Aktivität
und Energieverbrauch. Weitere Steigerung des Leptinspiegels durch Adipositas
bleibt ohne nennenswerte zusätzliche Wirkung. Für eine gewisse Zeit bestand die
Hoffnung, Leptin sei die Antwort für das verbreitete Übergewichtsproblem.
Tatsächlich hat ein sehr kleiner Prozentsatz adipöser Menschen einen
Leptinmangel. Die meisten stark übergewichtigen Menschen zeigen jedoch leider
eine zentrale Leptin-Resistenz (ähnlich der Insulin-Resistenz bei Diabetes mellitus Typ 2), d. h., sie
reagieren auf hohe Plasma-Leptin-Konzentrationen nicht mit vermindertem
Appetit.
"Hunger" entsteht im Hypothalamus
Das Gefühl, satt
zu sein, entsteht durch das Zusammenwirken von mehreren Signalen. Eine wichtige
Rolle spielt dabei eine zentrale Kooperation von Leptin und Insulin. Beide
stimulieren POMC (Pro-Opiomelanocortin)-exprimierende Neuronen im Nucleus arcuatus. Diese anorexigenen
(appetithemmenden) Neuronen sezernieren in ihren Synapsen im Nucleus paraventricularis ein
POMC-Fragment, α-MSH (α-melanocyte
stimulating hormone), das bestimmte Melanocortin-Rezeptoren (MC4R) auf den
dortigen "Sattheitsneuronen" aktiviert und eine Einstellung der
Nahrungsaufnahme sowie eine Erhöhung der Stoffwechselrate zur Folge hat. Etwa
4% der Patienten mit schwerer kindlicher Adipositas zeigen Mutationen im MC4R.
POMC-Neuronen sezernieren überdies CART (Protein des cocaine-
and amphetamine-regulated transcript), ein Neuropeptid, das einen G-Protein gekoppelten Rezeptor aktiviert, auch Sattheit vermittelt und die
Aktivität steigert. Die Aktivität der POMC-Neuronen wird übrigens auch durch
Nikotin gesteigert, was den niedrigeren durchschnittlichen body mass index von Rauchern erklärt sowie die Tendenz zur
Gewichtszunahme nach Rauchentwöhnung.
Pharmakologische Querverstrebung: Medikamente aus der Amphetaminfamilie wurden immer
wieder als Appetitzügler eingesetzt. Diese Praxis wurde in der EU allerdings weitgehend
beendet. Manche Präparate wiesen Suchtpotential auf, andere hatten zu viele
unerwünschte Wirkungen: Herzrasen, Stimmungsschwankungen, Verstopfung. Mechanismus ist eine Steigerung der Konzentration von
Serotonin und Noradrenalin im synaptischen Spalt, da POMC-Neuronen auch über
Serotonin- 5‑HT2C-Rezeptoren aktiviert werden. Die gleichzeitige
Aktivierung von 5-HT2B fördert allerdings die Fibrosierung von
Herzklappen und Pulmonalarterienwand und führte immer wieder zu pulmonalem
Hochdruck und Klappenproblemen. Der 5‑HT2C–Agonist Lorcaserin
war bis 2020 in den USA, nicht aber in der EU zur Unterstützung der Gewichtsabnahme
zugelassen. Ebenso ist die Kombination Phentermin
(Amphetaminderivat)/Topiramat (Antiepileptikum) in den USA, nicht jedoch in der
EU zugelassen. In der EU zugelassen ist die Kombination Bupropion/Naltrexon, die bei der Testung gegenüber Placebo nur eine
Gewichtsabnahme von 3-5% zeigte. Bupropion aus der Amphetamingruppe wird als
Einzelsubstanz zur Raucherentwöhnung und zur Behandlung schwerer Depressionen
eingesetzt, Naltrexon, ein Opioidrezeptorantagonist, zur Rückfallsprophylaxe
ehemals Opioid‑ und Alkoholabhängiger. Naltrexon hemmt eine β‑endorphinvermittelte
Autoinhibition der POMC-Neuronen. Beide haben als Nebenwirkung Appetithemmung,
die nun im Kombinationspräparat zur Hauptwirkung erklärt wurde. Zu den sehr häufigen
Nebenwirkungen gehören Übelkeit, Erbrechen, Verstopfung und Bauchschmerzen, was
eine Appetithemmung durchaus plausibel erscheinen lässt. In der klinischen
Testung brachen 24% der Probanden die Behandlung aufgrund eines unerwünschten
Ereignisses ab.
Leptin hemmt
dagegen eine zweite Gruppe von Neuronen im Nucleus
arcuatus, die Agouti-related peptide (AgRP)
und neuropeptide Y (NPY) freisetzen. Diese
wirken den POMC-exprimierenden Neuronen entgegen. AgRP wirkt am MC4R
antagonistisch. Die AgRP-"Hungerneuronen" fördern Hungergefühl, Nahrungsaufnahme und Aufbau
von Depotfett, während sie die Bereitschaft zu Aktivität und die Thermogenese
im braunen Fettgewebe drosseln. Ein eindrucksvolles Experiment, um die
AgRP-Neuronen persönlich im vollen Schwung zu erleben, besteht darin, 24
Stunden lang keine Kalorien zu sich zu nehmen und sich dabei zu beobachten:
antriebslos, frierend, übelst gelaunt.
Doch dann die
Belohnung: ein gutes Essen! Schon die Aussicht auf baldige Nahrungsaufnahme hemmt über
ZNS-Verknüpfungen die Aktivität der AgRP-Neuronen. Unser ZNS ist ein Organ zur
Berechnung der Zukunft: Sitzen wir vor dampfenden Tellern, steigt unsere
Zufriedenheit sofort und der Hunger lässt bereits nach; wir müssen nicht unter
bohrenden Unlustgefühlen vor uns hin löffeln, bis erst nach dem Essen Leptin
und Insulin ansteigen.
Was ist Hunger?
Hunger ist ein zunächst ungerichtetes Gefühl der Unlust, das durch die
Aktivierung von AgRP-Neuronen produziert wird. Wir versuchen, diesen unangenehmen
Zustand zu vermeiden und lernen früh, welche Verhaltensweisen
(Nahrungsaufnahme) die AgRP-Neuronen zum Schweigen bringen. Babys und kleine Kinder zeigen ihr
Unlustgefühl bei Hunger sehr direkt. Sie können sich noch nicht selber helfen,
doch haben sie keinerlei Schwierigkeiten, die Eltern entsprechend zu
konditionieren.
Sensorische Einflüsse (der dampfende Teller) reduzieren die Aktivität der AgRP, aber eine weitere frühe Dämpfung geschieht in einer zweiten Phase durch Signale aus dem Darm. Das hat wahrscheinlich folgenden Hintergrund: würden wir essen, bis Glucose, Leptin und Insulin hoch genug sind, um AgRP-Neuronen abzuschalten, würden wir über das Ziel hinausschießen, da ja dann noch eine große Menge Nahrung im Darm zur Absorption ansteht. Also meldet der Darm bereits früher eine Schätzung, dass nun wohl mit einer gewissen Menge an Nahrung gerechnet werden kann ("Darm an Großhirn! Darm an Großhirn!"), und dass das Hungergefühl nun bitte aufgehoben werden möge. Dies geschieht über Signalpeptide wie PYY und GLP-1, die einerseits über afferente Neuronen des autonomen Nervensystems (N. Vagus zu Nucleus tractus solitarii), andererseits direkt ans ZNS melden.
PYY wird nach der Nahrungsaufnahme durch neuroendokrine L-Zellen in der Schleimhaut des distalen Darms freigesetzt. PYY-Spiegel steigen innerhalb von 15 min an, lange vor Nahrungsmittel die L-Zellen erreichen, und bleiben bis zu sechs Stunden erhöht. Vergleicht man Mahlzeiten gleichen Brennwerts mit jeweiliger Betonung auf Kohlenhydraten, Protein oder Fett, so findet man die höchsten Plasmaspiegel von PYY nach proteinreichen Mahlzeiten. Abgesehen von peripheren Effekten, wie die Reduktion von Magenmotilität und –sekretion, fördert PYY zentral das Sättigungsgefühl. PYY stimuliert Y2-Rezeptoren im Hypothalamus. Das führt wieder zur Aktivierung der appetithemmenden POMC-exprimierenden Neuronen im Nucleus arcuatus. Zusätzlich zu PYY trägt auch Cholezystokinin zur Entstehung des postprandialen Sättigungsgefühls bei.
Praktische Bedeutung: Eine proteinreiche Mahlzeit verzögert das Wiederauftreten des Hungergefühls am effektivsten und erleichtert es damit, eine generelle Kalorienreduktion durchzuhalten.
Protein wird einerseits täglich benötigt, andererseits gibt es davon keine Speicherform. Die Proteinaufnahme könnte damit ein sinnvoller Parameter zur Regelung der Nahrungsaufnahme sein. Die protein leverage hypothesis postuliert, dass man so lange isst, bis man genug Protein aufgenommen hat. Isst man also kohlenhydrat- und fettreiche Nahrung, hat man nach dieser Hypothese so lange weiter Appetit, bis man die benötigte Proteinmenge erreicht.
Glucagon-like peptide‑1 (GLP‑1)
Ein zweites von
intestinalen L-Zellen in Ileum und Kolon freigesetztes Hormon ist GLP‑1.
Das Peptid aus 31 Aminosäuren hat eine Plasmahalbwertszeit von nur 2 Minuten,
da es rasch durch die Protease DPP4 (Dipeptidylpeptidase 4) gespalten wird; es
ist daher als Medikament ungeeignet. DPP‑4 wird auf den meisten Zellen
als Transmembranprotein CD26 exprimiert, aber auch in löslicher Form
sezerniert. Es spaltet Dipeptide (X-Prolin oder X-Alanin) vom N-Terminus von
Polypeptiden. GLP‑1 verstärkt die glucoseabhängige Insulinsekretion durch
β-Zellen im Pankreas, vergrößert die β‑Zellmasse und hemmt die
Glucagonfreisetzung. Es verzögert die Magenentleerung und verstärkt zentral das
Sättigungsgefühl; Patienten, die GLP-1-Agonisten anwenden, berichten, dass ihre
Hungerattacken ausbleiben und dass ihre Aufmerksamkeit nicht mehr, wie vorher, häufig
um das Essen kreist ("food noise").
Die durch GLP‑1-angesprochenen Neuronen sitzen nicht nur im Hypothalamus,
sondern auch in anderen Hirnregionen, darunter die Area postrema (zirkumventrikuläres Organ am caudalen Ende der
Rautengrube, dorsal des Nucleus tractus
soltarii).
Pharmakologische Querverstrebung: Zwei Klassen von Medikamenten werden eingesetzt, um die GLP‑1-Wirkung bei Diabetikern zu verstärken:
- GLP‑1-Agonisten mit längerer Halbwertszeit wie Liraglutid, Semaglutid
- DPP4-Inhibitoren wie Sitagliptin
Da Diabetes-Patienten unter der Therapie mit GLP-1-Agonisten deutlich Gewicht verloren, wurden diese zusätzlich zur Unterstützung der Gewichtsabnahme bei Adipositas zugelassen.
Semaglutid führte zu 17%
Gewichtsabnahme nach etwas mehr als einem Jahr, einmal wöchentlich subcutan
gespritzt. Es entfachte als Lifestyle-Medikament bei mäßig übergewichtigen Abnehmwilligen
derartige Nachfrage, dass es für die eigentlichen Diabetes-Patienten nicht mehr
ausreichend zur Verfügung stand. Gleichzeitig brachte es neue first world problems mit sich, wie das
"Ozempic face": Runzeln und
hängende Backen, da der Inhalt der Gesichtshaut nun geschrumpft ist – man wirkt
älter. Tragisch, nun benötigt man eine neue Runde Facelifting. Semaglutid ist
in 29 von 31 Aminosäuren identisch mit GLP‑1. Ein Ersatz von Alanin in der
zweiten Position verhindert die Spaltung durch DPP4, zusätzlich bewirkt ein
eingeführter Lipidrest eine Bindung an Albumin. Die Plasmahalbwertszeit steigt
damit von 2 Minuten auf 1 Woche. Zwei Dosierungen wurden eingeführt: Die
niedrigere Dosierung, Ozempic, hat
als Zielgruppe Typ 2-Diabetiker. In der höheren Dosierung, Wegovy, ist das Medikament zur Unterstützung der Gewichtsabnahme
zugelassen. Semaglutid muss vorsichtig eingeschlichen und langsam höherdosiert
werden, da es häufig unangenehme gastrointestinale Nebenwirkungen auslöst: in
der höchsten Dosierung Nausea in 44 %, Erbrechen 24 %, Diarrhoe 30 %,
Bauchschmerzen 20 %, Verstopfung 20 %. GLP-1-Agonisten sind eine Goldgrube für die
Hersteller: es bleibt bei einer Dauermedikation, sonst kommt das Gewicht wieder
zurück. Als Peptide müssen die ‑glutide injiziert werden, nicht wahr? Im
Prinzip, ja, doch erstaunlicherweise gibt es auch eine orale Version: in einer
höheren Dosierung mit spezieller galenischer Formulierung ist Semaglutid auch
in Pillenform – Rybelsus – für
Diabetes mellitus Typ 2 zugelassen. Dabei wird Semaglutid kombiniert mit einer
als SNAC abgekürzten lipidartigen Substanz. Beide gemeinsam werden bereits im
Magen transzytotisch über Vesikel durch die Magenschleimhaut transportiert. Die
Resorption ist aber nicht sehr effizient und verlangt eine Einnahme auf
nüchternen Magen mit wenig Wasser. Diese Darreichungsform benötigt daher
etwa zehnmal so viel Semaglutid wie die injizierbaren Formen, was beim
gegenwärtigen Semaglutid-Nachfrageüberhang kontraproduktiv ist.
Eleganter wäre es natürlich, den GLP‑1-Rezeptor
durch einen künstlichen kleinen, oral applizierbaren Agonisten zu aktivieren. Orforglipron, ein Nichtpeptid-Aktivator
des GLP-1-Rezeptors, in den USA bereits zugelassen, erfüllte diese Bedingungen.
Es ist bemerkenswert, dass es gelungen ist, einen Peptidrezeptor durch ein
kleines synthetisches und enteral resorbierbares Molekül zu aktivieren: Viele
Versuche bezüglich anderer Peptide und ihrer Rezeptoren sind bisher
gescheitert. Die Wirkung ist tendenziell etwas schwächer als jene der höchsten
Dosen der injizierbaren GLP‑1-Agonisten, die gastrointestinalen Nebenwirkungen
sind tendenziell etwas stärker. Die Pille gegen Übergewicht ist also – nach 60
Jahren vergeblichen Anläufen – Realität geworden.
Wenn der duale Rezeptoragonismus so gute Resultate
erzielt, warum nicht auf diesem Weg noch einen Schritt weitergehen? Der
Tripelrezeptoragonist Retatrutid aktiviert GLP‑1‑, GIP‑ und Glucagonrezeptoren (triple G-agonist) und befindet sich in
fortgeschrittenen Phase 3-Studien. Aktivierung des Glucagonrezeptors
erscheint zunächst kontraproduktiv: Glucagon, das uns über den Zeitraum helfen
soll, in dem wir gerade nichts zu essen haben, steigert den Blutzucker. Glucagon
steigert aber auch die Lipolyse, was ja bei Übergewicht sehr erwünscht ist. In Phase 2-Studien ergab sich nach 48 Wochen ein Gewichtsverlust bis zu 24%
gegenüber 2% für Placebo, nach 68 Wochen 28%. Besonders attraktiv ist die Beobachtung, dass die
Leberverfettung in einem hohen Prozentsatz der Behandelten zurückging.
Der Gewichtsverlust unter Therapie mit GLP-1-Agonisten bedeutet leider nicht nur eine Abnahme von Fett, sondern zu einem kleineren Teil auch die Abnahme von Muskelmasse. Es ist daher umso wichtiger, dass die Betroffenen ihren Lebensstil umstellen und diesem unerwünschten Effekt durch körperliches Training entgegenwirken. GLP-1-Agonisten lösen häufiger als Placebo akute Pankreatitis, Ileus und die Bildung von Gallensteinen aus.
Ghrelin wird als der Haupt-Gegenspieler
von Leptin betrachtet. Es wird während der Hungerphase durch bestimmte
Schleimhautzellen des Magens, besonders im Fundus, gebildet, sowie im
Pankreas. Bei der
adipositaschirurgischen Methode der Sleeve-Gastrektomie wird ein Großteil des
Fundus entfernt und damit auch die Ghrelinproduktion stark reduziert.
Ghrelin könnte der Name eines bösartigen Hungerkobolds sein, doch prosaischerweise ist es lediglich ein Akronym für growth homone release-inducing, da es auch an der Freisetzung von
Wachstumshormon beteiligt ist. Es ist ein Peptid von 28 Aminosäuren. Sein Rezeptor wird
einerseits von zentralen Neuronen im Nucleus
arcuatus, andererseits von peripheren afferenten Vagus-Neuronen exprimiert.
Ghrelin steigert das Hungergefühl und den Appetit. Es wird durch sportliche
Anstrengung unterdrückt. Zusammenfassend trägt Ghrelin zur Steigerung des Körpergewichts
und zum Längenwachstum bei.
6. KOHLENHYDRAT-VERDAUUNG UND AUFNAHME
Einfachzucker und raffinierter Zucker (Saccharose) in Form von Schokolade, Keksen, Konditoreiwaren etc. haben auf die meisten von uns eine große Anziehungskraft. Supermärkte steigern ihre Umsätze, indem sie diese regalweise dort positionieren, wo wir vor der Kasse stauen. Süßes wirkt positiv auf unser ZNS-Belohnungszentrum. Für unsere Vorfahren im altsteinzeitlichen Afrika war das ein nützlicher Mechanismus. Süßem begegneten sie selten, vielleicht, wenn es Beeren gab, oder wenn sie einen Bienenstock fanden. Denken wir an Weintrauben: Sie enthalten zur Hälfte Glucose (Traubenzucker), zur Hälfte Fructose, die gar nicht verdaut, sondern nur resorbiert werden müssen. Sie lösen eine enorme Insulinantwort aus und eignen sich damit hervorragend, in guten Zeiten Fettdepots für schlechte Zeiten anzulegen. In unserer Situation der dauernden reichlichen Verfügbarkeit führt das zu Übergewicht, metabolischem Syndrom und DM2.
Der Großteil natürlicher
Kohlenhydrate wird in Form von Stärke aufgenommen. Stärke besteht aus
geschichteten, wasserunlöslichen Körnern aus im Mittel 20-30 % Amylose und
70-80 % Amylopektin. Getreidekörner enthalten etwa 75 Gewichtsprozent
Stärke, Reis 90 %, die wasserhaltige Kartoffel 15 %.
Amylose besteht aus
linearen Ketten von α-1,4-glycosidisch verknüpften Glucose-Einheiten –
1000-2000 bei Getreide, 2000 bis 4500 bei Kartoffeln.
Amylopektin ist im
Grunde gleich aufgebaut wie unser Glykogen: aus α-1,4-glycosidisch verknüpften Glucose-Einheiten
plus α-1,6-glycosidischen
Verzweigungen. Ein Amylopektin-Makromolekül ist aber viel größer als ein
Glykogenmolekül und etwa 100 mal so groß wie Amylose. Die Ketten liegen, wie
die Speichen in einem zusammengeklappten Schirm, eng aneinander und bilden so
eine semikristalline Struktur.
Durch die
Wasserunlöslichkeit der Stärkekörner sind diese für uns kaum verdaubar: die
α‑Amylase kann nur ein wenig an der Oberfläche des Partikels
knabbern, das Stärkekorn verhält sich als Ballaststoff. Das ist z.B. der Fall,
wenn wir Rohkost essen. Es war deshalb eine glänzende Idee einer besonders intelligenten
Urahnin, deren Namen leider vergessen ist, sich das Feuer nutzbar zu machen und
eine ebenso glänzende Idee von WilMa Feuerstein
(WMF), den Kochtopf zu entwickeln. Wenn wir die Rohkost in Wasser zu Suppe
erhitzen, sprengen wir die Stärkekörner auf und machen deren Glucoseketten der
α‑Amylase zugänglich. Damit holen wir aus denselben Pflanzen ein
Vielfaches des Rohkostnährwerts. Leider ist dieser Prozess bis zu einem
gewissen Grad umkehrbar. Steht die hydrierte Stärke eine gewisse Zeit kühl,
bildet sich die semikristalline Struktur wieder aus. Wir nennen das "Retrogradation".
Retrogradation macht unser Brot altbacken: es verliert Wasser und wird härter.
Besonders rasch geht das um 0°C, darum sollte man Backwaren nie im Kühlschrank
aufbewahren. Die Verhärtungswirkung lässt sich durch Backzusätze verzögern:
Fette, Emulgatoren und Enzyme, insbesondere wieder Amylasen, welche die Stärke
bereits vor dem Backprozess verkleinern sowie Lipasen, die aus Fetten
eine Fettsäure abspalten und damit wieder Emulgatoren erzeugen.
Wie gut wir Stärke
verdauen können, hängt also von der Zubereitung der Lebensmittel ab. Nicht im
oberen Dünndarm verdaubare Stärkeanteile nennen wir resistente Stärke:
RS1 – physisch weggeschlossene
Stärke, wie in unverarbeiteten Körnern
RS2 – auf Grund ihrer
Struktur für Amylase nicht zugänglich, wie in Rohkost
RS3 – wegen
Retrogradation nicht mehr zugänglich
RS4 – neu vernetzte
Stärkeabbauprodukte wie resistente Dextrine
RS5 – mit Lipiden komplexierte Stärken
Resistente Stärkeanteile sind für uns nicht vollständig verloren: Sie gelangen in den Dickdarm und dienen dort als Futter für viele Bakterien, die sie zu kurzkettigen Fettsäuren wie z.B. Butyrat verstoffwechseln. Kurzkettige Fettsäuren werden im Dickdarm absorbiert; außerdem ernähren sie dort direkt unsere Enterozyten.
Die Verdauung von Amylose und Amylopektin beginnt
mit dem Enzym α-Amylase, das in Speichel und Pankreassekret vorkommt.
Dazwischen, im Magen, pausiert die Kohlenhydratverdauung, da die α-Amylase
durch die Magensäure inaktiviert wird. α-Amylase ist prinzipiell
in der Lage, α‑1,4‑glykosidische Bindungen zwischen
Glucoseeinheiten zu spalten, jedoch keine endständigen und keine, die
α‑1,6‑glykosidisch verbundenen Einheiten benachbart
sind. Aus der α-Amylase-Verdauung resultieren also keine Monosaccharide,
sondern Zweier- (Maltose) und Dreier- (Maltotriose) Einheiten von Glucose
sowie die Verzweigungsstücke um α‑1,6‑verbundene Einheiten,
die sogenannten Limit-Dextrine.
Die nächste Phase der Kohlenhydratverdauung
erfolgt durch die drei Typen von Bürstensaumenzymen: Maltase (Glucoamylase),
Lactase und Sucrase-Isomaltase. Maltase spaltet Maltose, Maltotriose
und noch etwas längere 1,4‑verbundene Polymere in ihre Glucose-Einheiten.
Lactase spaltet das Disaccharid Laktose (Milchzucker) in seine Monosaccharid-Einheiten
Glucose und Galactose. Sucrase-Isomaltase ist ein zusammengehängtes
Doppelenzym. Die Sucrase-Einheit spaltet das Disaccharid Saccharose
(den Zucker aus der Zuckerdose) in seine Monosaccharide Glucose und
Fructose. Die Isomaltase-Einheit spaltet die α‑1,6‑glykosidische
Bindung der Limit-Dextrine und ist ebenfalls in der Lage, 1,4‑verbundene
Einheiten zu spalten.
Die resultierenden Monosaccharide
werden schließlich durch Transporter in die Enterozyten aufgenommen.
Glucose wird zusammen mit Na+ im Verhältnis 1:2 durch den
Natrium/Glucose-Cotransporter 1 (SGLT1) importiert. Bei mäßigem, oral
behandelbarem Volumen- und Elektrolytverlust empfiehlt es sich daher,
nicht nur eine Elektrolytflüssigkeit zu geben, sondern Kohlenhydrate
oder Glucose zuzusetzen, da die Na+-Aufnahme so beschleunigt
wird. SGLT1 kann Glucose und Galactose über die apikale Enterozytenmembran
schleusen, nicht aber den Fünferring der Fructose. Fructose wird über
GLUT5 aufgenommen. Alle drei Monosaccharide verlassen den Enterozyten
auf der basolateralen Seite über GLUT2.
Glykämischer Index
Der Großteil der mit der
Nahrung aufgenommenen Kohlenhydrate erscheint im Blut in der Form von
Glucose. Wie rasch das geschieht und wie hoch die Glucosespitze tatsächlich
wird, hängt von der konkreten Zusammensetzung der Mahlzeit ab. Der glykämische
Index (GI, Glyx) ist der Versuch, diese Eigenschaft von Nahrungsmitteln
mit Hilfe einer einfachen Zahl auszudrücken. Der glykämische Index eines
Nahrungsmittels ist definiert als das Verhältnis des Integrals der Blutzuckerkurve
(area under the curve) über zwei Stunden
nach Verzehr jener Menge des Nahrungsmittels, die 50 g Kohlenhydrate
enthält, zum Integral der Blutzuckerkurve von 50g Glucose, multipliziert
mit 100. Glucose selbst hat damit einen GI von 100. Nahrungsmittel mit hohem glykämischem Index, wie Weißbrot, Nudeln, Kartoffeln,
die meisten Reissorten und die typischen industriellen Frühstücksflocken liegen
um 70 und darüber; Nahrungsmittel mit niedrigem GI wie Gemüse und das meiste
Obst, Vollkornprodukte sowie Molkereiprodukte liegen um 55 und darunter. (Achtung:
in manchen, vor allem US-amerikanischen Tabellen, wird statt Glucose
Weißbrot als Standard verwendet, was zu anderen Zahlen führt). Wir müssen uns allerdings bewusst sein, dass die Blutzuckerkurve nicht nur durch das Nahrungsmittel determiniert wird, sondern auch durch individuelle Eigenschaften des Essers - beispielsweise durch individuelle Variation der Verdauungseffizienz oder durch allfällige Insulinresistenz. Dem GI
wird in vielen populären Diäten Bedeutung beigemessen, z. B. in der
Glyx-Diät, der South Beach-Diät etc. Ist das berechtigt?
Die Aufnahme von Nahrungsmitteln
mit hohem GI führt zu einem raschen Anstieg des Blutzuckers, gefolgt
von einem entsprechenden Anstieg des Insulinspiegels. Dadurch wird in
den nächsten Stunden die Verbrennung von Kohlenhydraten zu Lasten der
Verbrennung von Fett gefördert, während überschüssige Glucose zu Fett
umgewandelt wird. In sorgfältig kontrollierten Tierversuchen, z. B.
mit Ratten, setzten Tiere unter hoher GI-Diät wesentlich mehr Fett an
als Tiere, die mit einer niedrig-GI-Diät gleichen Energiegehalts und
gleicher Nahrungszusammensetzung, inklusive gleicher Kohlenhydratmenge,
ernährt wurden. Im Gegensatz zu den niedrig-GI-Tieren entwickelten die
hoch-GI-Tiere auch Insulinresistenz.
Ob diese Resultate auf
den Menschen übertragbar sind, ist in der wissenschaftlichen Literatur
heftig umstritten. Am Menschen können diese Experimente nicht mit derselben
Rigorosität durchgeführt werden. Veränderungen am GI einer komplexen
Diät ändern notwendigerweise auch Faktoren wie Ballaststoffgehalt, "Biss",
Zuckergehalt und damit deren Schmackhaftigkeit. Wir alle tendieren dazu,
mehr zu essen, wenn es uns schmeckt. Bestimmend für die Entwicklung
von Übergewicht bleibt die positive Energiebilanz; wenn überhaupt, spielt
der GI eine dieser Bilanz nachgeordnete Rolle. Um auf der sicheren Seite
zu sein, erscheint es bis zur endgültigen Klärung trotzdem sinnvoll,
in unserer Ernährung Nahrungsmittel mit niedrigem GI zu betonen.
Praktische Umsetzung: Eine Umstellung auf niedrigen GI erreicht
man beispielsweise beim Frühstück, indem man Weißbrot mit Marmelade oder industielle Frühstücksflocken
durch Vollkornmüsli mit Joghurt, frischen Früchten und Nüssen ersetzt.
Laktose-Intoleranz
Lactase ist eine der Disaccharidasen
am Enterozyten-Bürstensaum. Sie spaltet das Disaccharid Laktose in die
Monosaccharide Glucose und Galactose. Während das Enzym in Säuglingen
und Kleinkindern stark exprimiert wird, lässt die Expression im Lauf
des Lebens nach. Deswegen tendieren Erwachsene weltweit dazu, im Lauf
ihres Lebens Laktose-intolerant zu werden. Eine Ausnahme bilden die
Nachkommen von Populationen, die sich in ihrer Ernährung traditionell
stark auf Milchprodukte stützen, wie in Nordeuropa. Diese Art der Ernährung
übte einen Selektionsdruck aus, der Individuen bevorzugte, die in der
Lage waren, die Expression des Enzyms länger aufrecht zu erhalten. Diese Fähigkeit beruht auf einem einzigen SNP 14.000
Basen upstream des Laktasegens:
normalerweise befindet sich dort ein C, bei Nachkommen der nordeuropäischen
Milchbauern erzeugt ein T eine Bindungstelle für den Transkriptionsfaktor Oct1.
Dieser zusätzlich gebundene Transkriptionsfaktor führt zur anhaltenden Expression des
Laktasegens. Laktose-"Intoleranz" ist also der Normalfall,
Laktasepersistenz die Ausnahme. Das erklärt, weshalb Laktose-"Intoleranz" bei
Menschen mit afrikanischer oder asiatischer Abstammung die Regel ist, aber
selten in Skandinavien vorkommt. Innerhalb Europas wird die
Laktose-"Intoleranz" von Norden nach Süden häufiger; in den
deutschsprachigen Ländern sind etwa 15% betroffen, in den Mittelmeerländern
sind die Prozentsätze wesentlich höher.
Wenn Laktose nicht gespalten
wird, kann sie von den Enterozyten nicht resorbiert werden. Durch den
osmotischen Effekt der Laktose, sowie durch sekundär veränderte Zusammensetzung
der Darmflora leiden betroffene Individuen unter Diarrhoe und Flatulenz
nach dem Genuss von Milchprodukten (Milch, Eis, Käse, Schokolade
). Da Darmbakterien bei der Metabolisierung von Laktose H2 produzieren, kann ein Atemtest auf H2 nach einer Testdosis von
Laktose zur Diagnose verwendet werden.
Die Kapazität des Fructose-Transporters GLUT5 ist begrenzt und variiert von Individuum zu Individuum. Wird die Transportkapazität überschritten, verursacht Fructose durch osmotischen Effekt und bakterielle Verstoffwechselung wiederum Diarrhoe und Flatulenz. Reich an Fructose sind der Zucker in unserer Zuckerdose, Früchte, Honig und mit Maissirup industriell hergestellte Lebensmittel. Ein hoher Prozentsatz der Menschen europäischen Ursprungs leidet an dieser Fructose-Malabsorption. Davon zu unterscheiden ist die sehr seltene hereditäre Fructose-Intoleranz, die durch eine Defizienz des Enzyms Aldolase B verursacht wird. Aldolase B ist das Enzym, das Fructose in der Leber in zwei Moleküle zu je drei Kohlenstoffatome spaltet.
Durch die Beliebtheit von
raffiniertem Zucker (Saccharose), die Verwendung von Glucose-Fructose-Sirup (Maissirup mit
hohem Fructosegehalt) in der industriellen Nahrungsmittelherstellung
und die Zugabe von Fructose zu vielen Fertiggetränken weist unsere westliche
Ernährung einen hohen Fructoseanteil auf. Fructose wird nach der Aufnahme
im Darm von der Leber quantitativ aus dem Blut extrahiert und metabolisiert.
Das dazu benötigte ATP wird dabei zu AMP und zum Teil weiter zu Harnsäure
abgebaut. Es gib Hinweise, dass hoher Fructosekonsum so die Entstehung von Gicht begünstigt.
Ein Teil der von der Leber metabolisierten Fructose wird in der Form von
Glucose und Laktat ins Blut abgegeben, ein Teil zur Fettsynthese verwendet, was non-alcoholic fatty liver disease begünstigt. Bei Nagern führt fructosereiche Ernährung
zu Insulinresistenz. Eine lange wissenschaftliche Debatte wird darüber
geführt, ob das auch bei den tatsächlichen Fructosemengen, die der Mensch
konsumiert, der Fall ist. Während es möglich erscheint, das Fructose
auf Grund seiner ausschließlichen Verstoffwechselung in der Leber eine
hepatische Insulinresistenz begünstigt, ist eine solche wahrscheinlich
nur ein Teilaspekt eines allgemeineren Problems: wir nehmen zu viel
einfache Zucker zu uns, und das führt zu Gewichtszunahme, Lipotoxizität
und Insulinresistenz.
Eine glucosebetonte Mahlzeit löst in β-Zellen des Pankreas Insulinsekretion aus. Bei Fructose ist dieser Effekt viel geringer, da wegen seiner niedrigen Plasmaspiegel (<0,5 mM, verglichen mit 5,5 mM = 100 mg/dl für Glucose) viel geringere Mengen durch GLUT2 in die β-Zelle transportiert werden, sodass der Insulinspiegel nach Fructosezufuhr niedriger bleibt. Das klingt auf den ersten Blick wünschenswert, denn Insulin wandelt ja Kohlenhydrate in vergrößerte Fettspeicher um. Nun haben wir aber gerade gesehen, das Fructose ganz ohne die Notwendigkeit von Insulin zu vergrößerten Fettspeichern umgewandelt wird. Erinnern wir uns, dass Insulin, zusammen mit Leptin, im Hypothalamus als Sättigungssignal wirkt. Konsumiert jemand Fructose, fühlt sie sich weniger gesättigt als eine Vergleichsperson, die eine isokalorische Menge Glucose zu sich genommen hat. Experimentell wurde nachgewiesen, dass die Esslust derselben Personen nach der Einnahme einer fructosehaltigen Limonade höher war als nach einer glucosehaltigen. Fructose könnte damit unser generelles Problem, zu viel einfache Zucker zu uns zu nehmen, noch akzentuieren, das zu Übergewicht, Lipotoxizität und Insulinresistenz führt.
Acrylamid
Acrylamid entsteht durch Rösten oder Backen
stärkehältiger Lebensmittel wie Chips, Keksen, Pommes frites oder Kaffee. Es
wird mit der Nahrung aufgenommen. In der Leber entsteht daraus mit Hilfe von
CYP2E1 Glycidamid, das DNA-Addukte bildet und so mutagen wirkt. Ob die Mengen
an Acrylamid in der Nahrung kritisch sind, ist umstritten. In einem
"umstrittenen" Fall ist es in der Regel wahrscheinlich, dass der
Effekt zumindest nicht sehr groß ist.
7. EISEN
Eisen ist in vielen biologischen
Systemen ein limitierender Faktor. Menschen wie Tiere benötigen Eisen,
um Sauerstoff zu transportieren und gezielt zu verstoffwechseln, in
Hämoglobin, Myoglobin sowie in den Zytochrom-Enzymen. Etwa zwei Milliarden
Menschen, mehr als ein Viertel der Weltbevölkerung, haben Eisenmangel;
etwa 500 Millionen leiden unter einer manifesten Eisenmangelanämie.
Eisenmangel ohne Anämie kann zu unspezifischen ZNS-Symptomen
(Müdigkeit, Konzentrationsschwäche, Kopfschmerzen, Restless Legs-Syndrom,
anderen Schlafstörungen, bei Kindern Verzögerungen der mentalen und motorischen
Entwicklung), Hautsymptomen (Mundwinkelrhagaden, Haarausfall) oder
Palpitationen führen. Auch Pilze und Bakterien benötigen Eisen und wenden mitunter viel Energie
und Ressourcen auf, um es zu bekommen. Ein Trick, mit dem unser Organismus
Infektionen bekämpft, ist, den Mikroorganismen das Eisen vorzuenthalten.
Eisen aus der Nahrung wird hauptsächlich im Duodenum absorbiert. In unserer Nahrung kommt es einerseits in eine Hämgruppe integriert als Häm-Eisen, andererseits frei vor. Der Großteil des Häm-Eisens stammt aus dem Myoglobin von Fleisch. Es wird zusammen mit der Häm-Gruppe durch einen speziellen Transportprozess aufgenommen und erst im Enterozyten freigesetzt. Dieser Prozess ist effizienter als der Transport freien Eisens. Freies Eisen liegt in der Nahrung dreiwertig (Fe3+) oder zweiwertig (Fe2+) vor. Fe3+ ist über einem pH von 3 nicht löslich; es formt stabile Komplexe mit verschiedenen Anionen und wird nicht resorbiert. Fe2+ ist bis zu einem pH von 8 löslich; es wird gemeinsam mit einem Proton durch den divalent metal transporter (DMT1) aufgenommen.
Ernährungsaspekte: 15%-35% des Hämeisens werden aufgenommen, weitgehend unabhängig von der Nahrungszusammensetzung. Dagegen werden nur 5-12% des Nicht-Hämeisens aufgenommen, und diese Aufnahme kann durch Nahrungsfaktoren weiter reduziert werden. Eisenbindende Phytate, z. B. in Getreide, Tannine, z. B. in grünem Tee, Schwarztee und Kaffee, oder der alkalische pH von Milchprodukten reduzieren diesen Prozentsatz. Aus diesem Grund geraten Veganer und Vegetarier leichter in einen Eisenmangel, besonders in Situationen erhöhten Bedarfs wie bei Kindern, Schwangeren, Stillenden und Frauen mit starker Menstruationsblutung. Um pflanzliches Eisen möglichst gut aufzunehmen, eignen sich Hülsenfrüchte (Sojabohnen, Linsen, Kichererbsen), Ölsaaten (Kürbiskerne, Sonnenblumenkerne), Nüsse (Mandeln, Walnüsse), Spinat (jaja, trotzdem), schwarze Melasse zeitlich getrennt von Tee- oder Kaffeegenuss. Zitronensaft (Ascorbinsäure, Säure) verbessert die Aufnahme, Protonenpumpenhemmer verschlechtern sie massiv.
Einschub Zink: Die in pflanzlicher Nahrung generell, aber in Getreide besonders
reichlich enthaltene Phytinsäure bindet nicht nur Eisen, sondern auch Zink.
Zugleich enthält pflanzliche Nahrung von vornherein weniger Zink als tierische.
Das macht Zink bei rein pflanzlicher Ernährung zu einem potentiell kritischen
Spurenelement. Zink ist für die korrekte Konformation vieler Proteine nötig (z.
B. Zinkfinger in Steroidhormonrezeptoren) sowie für die
enzymatische Funktion der meisten Metalloproteasen (z. B. Kollagenase).
Zinkmangel kann zu Wachstumsstörungen und Entwicklungsverzögerungen bei Kindern
führen, zu schlechterer Infektabwehr, Anorexie, psychiatrischen Störungen, Dermatosen,
brüchigen Nägeln, Haarausfall und verminderter männlicher Fruchtbarkeit.
Eisen wird durch den solute carrier Ferroportin aus dem duodenalen
Enterozyten ausgeschleust, ein Eisenexportprotein, das von allen Zellen
exprimiert wird. Da Fe2+ wegen seiner Fähigkeit, Hydroxylradikale
zu generieren, gefährlich ist, wird es während des Ausschleusungsvorgangs
durch das Kupfer-Protein Hephaistin in dreiwertiges Eisen übergeführt.
Als Fe3+ wird
es dann an Transferrin gebunden im Blut transportiert. Auch im Blut
gibt es ein Kupferprotein, Coeruloplasmin, das zweiwertiges in dreiwertiges
Eisen überführen kann.
Wenn zelluläre Eisenspiegel
niedrig sind, wird die Aufnahme von Eisen aus dem Blut durch einen eleganten
Mechanismus angekurbelt. Die mRNA für den Transferrin-Rezeptor enthält
mehrere iron response elements
(IRE) in ihrer 3'‑untranslatierten Region (UTR), die charakteristische
stem-loop-Sekundärstrukturen bilden. Hat
die Zelle Eisenbedarf, werden diese Strukturen durch bestimmte Proteine,
IRE-binding proteins (IRE-BP) oder iron regulatory proteins (IRP), gebunden.
Diese Bindung in der 3'-UTR schützt die mRNA davor, durch RNasen abgebaut
zu werden, und sichert so eine hohe Expression des Transferrin-Rezeptors.
Sobald der intrazelluläre Eisenspiegel ausreichend angehoben ist, bindet
Fe2+ die iron regulatory
proteins und führt diese in eine Konformation über, die nicht mehr
an IRE binden kann. Dadurch wird die mRNA nun rasch abgebaut, sodass
weniger Transferrin-Rezeptormoleküle hergestellt werden und die Eisenaufnahme
in die Zelle reduziert wird.
Iron response elements werden auch umgekehrt verwendet: in der Ferritin-mRNA
ist ein einzelnes IRE in der 5'UTR vorhanden, kurz vor dem Translations-Initiationscodon.
Bei niedrigen intrazellulären Fe2+-Spiegeln wird das IRE
durch IRP gebunden und die Translation dadurch blockiert (die mRNA-Stabilität
wird dadurch offensichtlich nicht beeinflusst). Hohe intrazelluläre
Fe2+-Spiegel entfernen das IRP, sodass aktive Translation
große Mengen dieses Eisenspeicherproteins produziert. Der Großteil des
Ferritins befindet sich intrazellulär; Plasmaferritin, das sich im Gleichgewicht
mit intrazellulärem Ferritin befindet, kann jedoch Auskunft über die
Eisenspeicher des Körpers geben. Ferritin-Speicherung eines Eisenüberschusses
findet hauptsächlich in der Leber statt.
Die bis hier beschriebenen
Mechanismen sind zwar zunächst in der Lage, intrazelluläre Eisenspiegel
zu regulieren; durch dauernden Nachschub aus dem Darm würde der Organismus
aber mit der Zeit überladen werden. Daher ist die Leber der Ausgangspunkt
einer negativen Rückkoppelung. Hepatozyten "messen" den extrazellulären Eisenspiegel mit Hilfe eines Proteinkomplexes, der den Transferrin-Rezeptor
und das MHC-I-ähnliche Transmembranprotein HFE
(high Fe) enthält. Hinreichende Eisenspiegel führen zur Induktion und
Sekretion des Signalpeptids Hepcidin. Dieses Peptid aus 25 Aminosäuren
bindet an den Eisenexporter Ferroportin und führt zu dessen Internalisierung
und Abbau. Ist also ausreichend extrazelluläres Eisen vorhanden, blockiert Hepcidin damit die Freisetzung von Eisen
aus allen Zellen, inklusive aus den duodenalen Enterozyten. Nach kurzer
Zeit verhindert die Akkumulation von Eisen in der Duodenalzelle die weitere Eisenaufnahme aus dem Darm.
Wie wir bereits
in unserer Auseinandersetzung mit dem Immunsystem gesehen haben,
wird in einem zweiten regulatorischen
Mechanismus Hepcidin durch das Auftreten einer Entzündung reguliert.
IL‑6 und andere inflammatorische Zytokine induzieren bekanntlich
eine Akutphasenreaktion der Leber; diese beinhaltet auch eine Sekretion
von Hepcidin. In diesem Fall werden dadurch die Makrophagen, die viel
Eisen aus dem Abbau von alternden Erythrozyten, der Erythrophagozytose,
enthalten, dadurch gehindert, dieses Eisen abzugeben. In einem weiter
verstärkenden Mechanismus wird die Ferroportin-Expression der Makrophagen
direkt durch TLR4-Aktivierung sowie durch inflammatorische Zytokine
gebremst. Zusammen führen diese beiden Mechanismen dazu, dass Eisen
im retikuloendothelialen System sequestriert ("weggesperrt")
wird und damit den infizierenden Mikroorganismen nicht zur Verfügung
steht. Während einer akuten Infektion stellt dieser kurzfristige "interne
Eisenmangel" kein Problem dar. Bei einer chronischen Infektion,
wenn diese Sequestrierung über lange Zeit anhält, kann dieser Mechanismus
jedoch zu einer Eisenaushungerung der hämatopoetischen Vorläuferzellen
und damit zu einer "Eisenmangelanämie" trotz überladener retikuloendothelialer
Eisenspeicher führen.
Hämochromatose
Eisenüberladung wirkt bald
toxisch. Sie tritt sekundär nach häufigen Transfusionen oder bei bestimmten
hämolytischen Anämien auf, oder primär als hereditäre Hämochromatose.
Der Großteil dieser Patienten (type 1) hat eine Mutation im HFE-Gen, und hier wieder der Großteil
eine bestimmte Mutation, Cys282Tyr. Der Defekt führt über eine unzureichende
Hepcidin-Expression zu einer ungehemmten Eisenaufnahme aus dem Darm,
was im Lauf der Jahre zu einer Eisenüberladung des Organismus mit Leberzirrhose,
Diabetes mellitus, Arthritis und starker Hautpigmentierung führt. Über
Hepcidin wird der Gesamteisenspeicher bei gesunden Erwachsenen zwischen
2-6g reguliert; im Rahmen einer Hämochromatose wächst dieser jährlich
um 0.5 bis 1 g, und kann schließlich bis zu 50 g betragen. Die Symptome
der Erkrankung manifestieren sich daher bei Männern meist erst jenseits
des 40. Lebensjahres, bei Frauen durch die Monatsblutungen in der Regel
überhaupt nicht. Die Therapie ist mittelalterlich einfach, aber effektiv:
Aderlass, bis die Eisenspeicher korrigiert sind. Die C282Y-Mutation
ist erst vor 1200-1400 Jahren in Nordeuropa entstanden und im Verhältnis
dazu in Menschen mit europäischen Wurzeln sehr häufig: etwa 10% sind
heterozygot, ca. 1 Person in 200 bis 400 homozygot. Klinisch erkrankt
jedoch auch bei Männern nur ein Teil der Homozygoten. Die Häufigkeit
dieser jungen Mutation lässt einen selektiv wirkenden Vorteil vermuten.
Frauen könnte sie erleichtern, die monatlichen Eisenverluste auszugleichen.
Makrophagen, die bei Erkrankten eisenentleert sind, könnten in diesem
Zustand leichter mit intrazellulären Erregern wie Mycobacterium tuberculosis und Salmonella
typhimurium fertig werden.
8. FOLSÄURE
Folsäure kommt in vielen
Gemüsen vor, z. B. in Spinat, Salat, Bohnen, Broccoli; in Leber und
abgeleiteten Produkten sowie, wie dessen Anhänger selten zu betonen
vergessen, in Bier. Folsäure ist in der Tetrahydrofolat (THF)-form notwendig
für die Nucleotid-Synthese (sowohl für Purine, als auch für Thymidin) und damit für alle
Gewebe mit hoher Proliferationsrate. Zusätzlich wird es für viele C1-Gruppentransfers
mit Ausnahme von Carboxylierungen, die Biotin-abhängig sind, benötigt.
Ein typisches Beispiel ist die Synthese von Methionin aus Homocystein.
Homocystein ist toxisch; seine Konzentration wird durch diese
komplexe Reaktion mit Hilfe von THF und B12 niedrig gehalten. Folatmangel ist
die häufigste Ursache einer Hyperhomocysteinämie und daher ein kardiovaskulärer
und neurodegenerativer Risikofaktor. Abhängig von der spezifischen Reaktion, wird THF in einer von mehreren
Formen benötigt: die Synthese von dTMP aus dUMP ist z. B. auf 5,10‑methylen-THF
angewiesen, die Methioninsynthese auf N5‑methyl-THF.
Aus der Nahrung nehmen
wir Folat hauptsächlich in seiner Polyglutamatform auf. Im Duodenum
werden die Glutamatreste schrittweise von einer membranständigen Peptidase
abgespaltet. Monoglutamatfolat wird dann durch Austausch gegen ein Hydroxyl-Ion
(OH-) absorbiert. Folsäure wird hauptsächlich in der Leber
gespeichert. Sie wird zuerst zu Dihydrofolsäure (DHF), dann durch das
Enzym Dihydrofolatreduktase (DHFR) zur aktiven Tetrahydrofolsäure reduziert, um anschließend durch Serin, das
dadurch zu Glycin wird, mit der C1-Gruppe beladen zu werden.
Folsäuremangel führt zu
Problemen in allen rasch proliferierenden Geweben, wird aber klinisch
vor allem als Anämie sichtbar. Die Zahl der produzierten Erythrozyten
ist zu gering; in einem kompensatorischen Mechanismus werden sie dafür
mit möglichst viel Hämoglobin angestopft. Das macht sie allerdings wieder
anfälliger für den Abbau, da sie sich nicht so leicht durch die Spalträume
des Retikuloendothelialen Systems in der Milz quetschen lassen. Wir
sprechen von einer hyperchromen, makrozytären oder auch von einer megaloblastären
Anämie.
Zellproliferation ist besonders
hoch in der Embryonal- und Fetalperiode. Folsäuremangel in dieser Phase
kann zu Neuralrohrdefekten wie Spina
bifida führen. Schwangere werden aus diesem Grund routinemäßig mit
Folsäure substituiert. Da
Neuralrohrdefekte aber sehr früh in der Schwangerschaft auftreten, ist es wichtig,
schon vor der Empfängnis ausreichend Folsäure zu sich zu nehmen. Die Deutsche
Gesellschaft für Ernährung empfiehlt Folsäureeinnahme für "Frauen, die
schwanger werden wollen oder könnten".
9. VITAMIN B12
Eines der komplexesten
"kleinen" Moleküle unseres Körpers ist Vitamin B12 oder Cobalamin.
Bisher sind nur zwei Enzymreaktionen bekannt, für die dieses Koenzym
benötigt wird: die Synthese von Methionin aus Homocystein und die "Entzweigung"
von Methylmalonat, das beim Abbau ungeradzahliger Fettsäuren und Aminosäuren
entsteht. Bei der Methioninsynthese arbeitet B12 mit N5‑Methyl-THF
zusammen, von dem es die Methylgruppe übernimmt. Ohne B12 ist N5‑Methyl-THF
eine metabolische Sackgasse, aus der Tetrahydrofolat nicht mehr regeneriert
werden kann. Vitamin B12 kann nur von bestimmten Bakterien synthetisiert
werden, wird jedoch von vielen anderen Bakterien und allen Tieren, in Summe
also einer enormen Nahrungskette, gebraucht. Da es in Pflanzen nicht vorhanden
ist, müssen wir es mit tierischen Produkten aufnehmen: mit Fleisch, Fisch,
Eiern, und in einem geringeren Ausmaß auch mit Milchprodukten. B12-Mangel ist
daher noch vor Eisenmangel das Hauptproblem veganer Ernährung. Kein Problem ist
es, wenn man industriell mit Hilfe bestimmter Bakterien hergestelltes B12 als
Nahrungsergänzung zu sich nimmt. Für eine quantitativ ausreichende synthetische
Herstellung ist das Molekül zu kompliziert.
Vitamin B12 wird nur im Ileum absorbiert und muss bis dorthin vor Abbau im Verdauungstrakt geschützt werden. Im sauren Milieu des Magens wird es zunächst durch das Protein Haptocorrin (R-Protein) gebunden, das von Speicheldrüsen und Magenschleimhaut sezerniert wird. Parietalzellen der Magenschleimhaut produzieren gleichzeitig intrinsic factor (IF), ein Glycoprotein, das B12 später im Duodenum übernimmt, wenn Haptocorrin durch Pankreas-Proteasen abgebaut wird. Der B12:IF-Komplex ist in der tobenden Verdauungsschlacht außerordentlich stabil. Im Ileum angekommen, wird der Komplex über Rezeptor-vermittelte Endozytose aufgenommen. Dieser Prozess ist absolut IF-abhängig: freies B12 wird vom Rezeptor weder gebunden noch aufgenommen. Im Enterozyten dissoziiert der Komplex; B12 wird von Transcobalamin übernommen, das auch für die Exozytose und den Transport zur Leber sorgt. Ein großer Pool von Vitamin B12 besteht in der Leber und wird enterohepatisch rezirkuliert: mit der Galle ausgeschieden und im Ileum wieder resorbiert.
Wenn in einem der zahlreichen
Systeme, die zur Aufnahme von Vitamin B12 benötigt werden, ein Problem
besteht, wird zu wenig B12 resorbiert. Neben nahrungsbedingtem Mangel
bei strengen Vegetariern ist eine häufige Ursache ein Autoimmunreaktion
gegen Parietalzellen. Der Mangel wird schleichend wirksam, da in der
Leber eine mehrjährige Reserve besteht. Die Symptome sind die eines
Folsäuremangels. Fehlendes B12 blockiert auf die Dauer die Folsäurereserven
des Körpers in der N5‑Methyl-THF-Form, sodass zu wenig
5,10‑methylen-THF zur Verfügung steht. 5,10‑Methylen-THF
wird aber unbedingt für die Synthese von dTMP aus dUMP (Enzym: Thymidylatsynthase)
benötigt. Auch N10-Formyl-THF, das für die Synthese des Purin-Grundgerüsts
benötigt wird, ist zu wenig vorhanden. Dadurch ergibt sich dieselbe
Art von hyperchromer, makrozytärer Anämie wie bei Folsäuremangel. Es wäre auch möglich, diese Anämie durch dauernde Zufuhr von
frischer Folsäure zu kaschieren, doch wäre das ein Kunstfehler, da weitere
B12-Mangelsymptome durch Folsäurezufuhr nicht verhinderbar sind.
Weitere mögliche Symptome des B12-Mangels sind eine charakteristische atrophe Glossitis und neurologische Probleme, beginnend mit Polyneuropathie. In schweren Fällen ist auch das ZNS betroffen mit Erinnerungsstörungen, Depression, Schwäche und Ataxie. Für Schwäche und Ataxie sind hauptsächlich Entmarkungsherde in Rückenmarksbahnen verantwortlich, auf die sich der Name des Krankheitsbildes, funikuläre Myelose bezieht. Der pathophysiologische Mechanismus ist hier nicht im Detail geklärt, möglicherweise trägt ein erhöhter Homocysteinspiegel dazu bei.
10. SELEN
Betrachten wir Selen als
ein herausgegriffenes Beispiel für Spurenelemente. Selen steht mit Sauerstoff
und Schwefel in der sechsten Hauptgruppe; es reagiert also leicht und
nimmt gerne Elektronen auf, hat aber eine höhere Bereitschaft als Halogene,
diese auch wieder abzugeben. Es ist unabdingbar für einige wichtige
Redoxreaktionen, wie die Entgiftung von Peroxiden durch Glutathionperoxidasen,
sowie für die Aktivierung und Inaktivierung von Schilddrüsenhormon durch
Iodthyronin-Dejodinasen. Ein Beispiel dafür haben wir schon erwähnt: die
Aktivierung braunen Fettgewebes. Selen wird in Form einer besonderen Aminosäure,
der 21. Aminosäure, Selenocystein, in diese Enzyme eingebaut. In Selenocystein
nimmt Selen die Stelle des üblichen Schwefelatoms ein. Im genetischen
Code gibt es kein eigenes Codon für Selenocystein. Stattdessen kann
eines der drei Stop-Codons, UGA, mit Hilfe eines speziellen Translationsfaktors
als Selenocystein-Codon interpretiert werden, indem dieser an eine Signal-Schleifenstruktur
in der 3'‑UTR der Enzym-mRNA bindet.
In den meisten Gegenden
ist es nicht nötig, sich über Selen Gedanken zu machen. Es gibt allerdings
einige definierte Regionen, besonders in China, in denen der Boden wenig
Selen enthält und die Nahrungszufuhr daher zu gering ist. Selenmangel
kann dort zur Keshan-Krankheit führen, die durch eine lebensgefährliche
Kardiomyopathie gekennzeichnet ist.
Fettverdauung und
–aufnahme betrachten wir weiter unten im Rahmen der Leberfunktionen. Hier widmen
wir uns zunächst den Fettsäuren vom ernährungswissenschaftlichen Standpunkt
aus.
Docosahexaensäure ist
für die Entwicklung und Funktion von Gehirn und Auge wichtig; sie ist
angereichert in den Phospholipiden der Zellmembranen in Gehirn und Retina. DHA
macht 15‑20% aller Fettsäuren im frontalen Cortex aus. Da Veganer*innen
keine Fische essen, ist DHA ein potentiell kritischer Nährstoff für sie, speziell
wieder für Schwangere, Stillende und deren Kinder. Ein Ausweg besteht darin,
DHA an der Quelle noch vor den Fischen, aus Mikroalgenkulturen zu gewinnen und in Form von
Mikroalgenöl zuzuführen.
Es steht außer Zweifel,
dass das Gemisch an Fettsäuren, das wir mit der Nahrung aufnehmen, unsere
Gesundheit beeinflusst. Unser diesbezügliches Wissen stammt aus ernährungswissenschaftlichen
Studien, in denen der menschliche Organismus als black box behandelt wird: die Zusammensetzung
der Nahrung verschiedener Kohorten oder Populationen wird dabei korreliert
mit gemessenen Gesundheitsparametern. Die molekularen Mechanismen hinter
den Korrelationen bleiben dabei in der Regel im Dunkeln. Zwar wurden
zahlreiche Hypothesen formuliert, doch bleiben diese unzureichend geprüft.
Die Diskussionen über Fettsäuren
konzentrieren sich auf zwei Bereiche: n-3/n-6- und Trans-Fettsäuren.
n-3/n-6 mehrfach ungesättigte Fettsäuren
Abhängig von der Distanz
der ersten Doppelbindung vom Methylende der Fettsäure aus, also "vom
hinteren Ende", fallen die meisten mehrfach ungesättigten Fettsäuren
in eine von zwei Gruppen:
- n-3-Fettsäuren (häufig auch als ω-3-Fettsäuren
bezeichnet), wie z. B. α-Linolensäure (18:3), Eicosapentaensäure
(EPA; 20:5) und Docosahexaensäure (DHA; 22:6). Fettfische sind reich
an langkettigen n-3-Fettsäuren.
- n-6-Fettsäuren, wie Linolsäure (18:2) und Arachidonsäure
(20:4).
Ausgangspunkt war die Beobachtung,
dass Grönländische Inuit nur sehr selten koronare Herzkrankheit entwickelten,
obwohl sie sich praktisch ausschließlich von fettreicher tierischer
Nahrung ernährten. Da sie in Form von Fettfisch hohe Mengen langkettiger
n-3 PUFA zu sich nahmen, wurde ein kausaler Zusammenhang vermutet.
Seither durchgeführte Kohortenstudien sowie randomisierte Interventionsstudien
mit Fischöl führten im Einzelfall zwar zu widersprüchlichen Ergebnissen;
das Gewicht der Daten spricht jedoch für eine Bestätigung dieses Zusammenhangs.
Der
Mechanismus bleibt unklar. Beschrieben
wurde eine Hemmung der inflammatorischen Aktivität des Fettgewebes durch n-3
PUFA. Andererseits wurden auch für n-6-PUFA positive Effekte auf Arteriosklerose beschrieben. Wahrscheinlich
sind zwei Gesichtspunkte von Bedeutung:
1.
dass
wir ausreichend PUFA im Verhältnis zu gesättigten Fettsäuren aufnehmen
2.
dass
die aufgenommenen PUFA in einem günstigen Verhältnis n‑3:n‑6 liegen.
Empfohlen wird ein Verhältnis von mindestens 1:5; in unserer westlichen
Ernährung liegt das tatsächliche Verhältnis jedoch um 1:15.
Trans-ungesättigte Fettsäuren
Ungesättigte Fettsäuren
liegen in einer von zwei möglichen Konfigurationen vor: cis oder trans.
Cis bedeutet, das die Kohlenstoffketten auf derselben Seite der Doppelbindungsachse
liegen; die Fettsäure bekommt dadurch einen Knick. Dieser Knick hat
zur Folge, dass ein Bündel von Cis-Fettsäuren schwer zu "schlichten"
ist, während die kerzengeraden gesättigten Fettsäuren oder die nur ein
winziges Bajonett enthaltenden Trans-Fettsäuren viel leichter zu schlichten
sind. Mit anderen Worten: gesättigte und Trans-Fettsäuren, sowie komplexe
Lipide, die diese enthalten, haben einen relativ hohen Schmelzpunkt,
während Cis-Fettsäuren einen niedrigen Schmelzpunkt haben. So hat z.
B. die gesättigte C18-Fettsäure, Stearinsäure, einen Schmelzpunkt von
69°C, die mono-ungesättigte Trans-Fettsäure Elaidinsäure einen von 45°C,
während die entsprechende Cis-Fettsäure, Ölsäure, einen Schmelzpunkt
von nur 17°C hat. Bei einer Körpertemperatur von 37°C sind Lipide mit
cis-ungesättigten Fettsäuren daher "fluider" als solche mit gesättigten oder trans-ungesättigten
Fettsäuren, die eher dazu tendieren, sich zusammenzulagern. Mangelnde Fluidität
kann allerdings nicht die beobachteten negativen Effekte von Trans-Fettsäuren erklären,
da unser Organismus ja vowiegend gesättigte Fettsäuren enthält, die noch
weniger fluid sind. Diskutiert wird,
dass Trans-Fettsäuren durch ihre Bajonettstruktur Enzyme behindern, die andere Fettsäuren
metabolisieren sollen. Zwar ist man sich über die genauen Mechanismen noch
nicht einig, doch sind Trans-Fette mit einem höheren Risiko für Atherosklerose,
koronare Herzkrankheit und Schlaganfälle assoziiert. Zusätzlich wurde
beschrieben, dass Trans-Fette das unerwünschte LDL-Cholesterol heben und das
"gute" HDL-Cholesterol senken.
Also, wir wünschen mehr
Cis-Fette und weniger Trans-Fette. Woher kommen sie eigentlich?
Cis-Fette stellen den Großteil pflanzlicher Fette dar. Nach ihrer Extraktion liegen
sie durch ihren niedrigen Schmelzpunkt bei Raumtemperatur flüssig, als
Öle vor. Beispiele sind Olivenöl (75% Ölsäure, 10%
Linolsäure, 15% gesättigt), erucasäurearmes Rapsöl (60% Ölsäure, 20%
Linolsäure, 10% α-Linolensäure, 7% gesättigt, bis zu 4% Trans-Fette durch
den Raffinationsprozess), Maiskeimöl (50%
Linolsäure, 30% Ölsäure, 15% gesättigt), Sojaöl (55% Linolsäure, 25 Ölsäure,
10% gesättigt) und Sonnenblumenöl (65% Linolsäure, 20% Ölsäure, 10% gesättigt).
Eine Züchtung zur Reduktion der Linolsäure, high
oleic acid-Sonnenblumenöl (80% Ölsäure, 10% Linolsäure, 10% gesättigt) nähert
sich der Zusammensetzung von Olivenöl. Leinöl enthält als einziges Pfanzenöl vorwiegend n-3-Fettsäuren
(63% α-Linolensäure, 15% Linolsäure, 15% Ölsäure, 7% gesättigt). Es ist
besonders empfindlich gegenüber Sauerstoff, oxidiert leicht und wird dabei
rasch bitter. Tipp: In kleinen Mengen kaufen, großzügig als Salatöl verwenden
und rasch verbrauchen! Diese Prozentsätze (zusammengefasst in der Grafik nach dem nächsten Absatz) sind nur Anhaltspunkte,
da die Zusammensetzung erheblichen Schwankungen unterliegt.
Trans-Fette kommen in geringer Konzentration im Fett von Wiederkäuern vor. So kann Butter (70% gesättigt, 20-25% Ölsäure) bis zu 4% Trans-Fette enthalten. Aus den bisher erwähnten Gründen konsumierten
Menschen bis vor wenigen Jahrzehnten nur geringe Mengen an Trans-Fetten.
Das änderte sich mit der industriellen Nahrungsmittelproduktion. Pflanzliches
Öl ist billig zu gewinnen, doch oxidiert es leicht (es wird "ranzig"),
es ist schwerer zu transportieren und man kann es nicht aufs Brot streichen.
Also wurden Methoden entwickelt, Pflanzenfette zu härten (sprich: den
Schmelzpunkt hinaufzusetzen). Diese beruhen auf partieller Hydrierung.
Die Idee ist, einen Teil der Doppelbindungen mit Wasserstoff abzusättigen,
sodass teils gesättigte, teils ungesättigte Fettsäuren mit einer reduzierten
Anzahl an Doppelbindungen entstehen. Der Prozess hat jedoch den Nachteil,
dass er einen erheblichen Anteil der so behandelten Fettsäuren in die
trans-Konfiguration bringt. Im Grunde verwandeln wir damit gute Nahrungsfette
in schlechtere. Gehärtete Fette mit hohem Trans-Fett-Anteil verdrängten in der Folge andere
Fette in vielen Bereichen, speziell in der industriellen Herstellung
von Streichfetten, Backwaren, Knabbergebäck, Snacks und Fastfood.
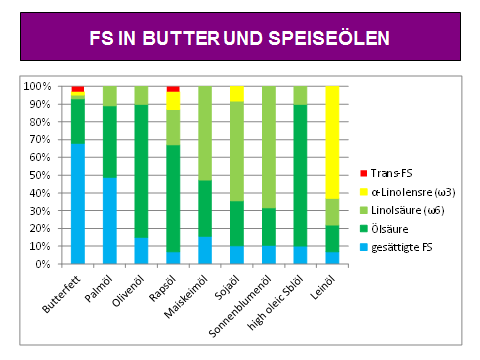
Den negativen gesundheitlichen
Aspekten von Trans-Fetten wurde über viele Jahre wenig Aufmerksamkeit
gewidmet. Später
führte wachsendes Gesundheitsbewusstsein allerdings in vielen Ländern zu Gesetzesänderungen,
welche den zulässigen Anteil der Trans-Fette in industriell hergestellter Nahrung
sowie in Restaurant-Speisen stark reduzierten.
Zur Reduktion kardiovaskulärer Erkrankungen empfiehlt die WHO seit 2018, nicht mehr als 10% des täglichen Kalorienbedarfs in Form von gesättigten Fetten und nicht mehr als 1% in Form von Trans-Fetten zu sich zu nehmen. Große epidemiologische Studien haben ergeben: Ersetzt man Kohlenhydratkalorien durch Fettkalorien, führt das zu einer gesenkten Mortalität, solange es sich um ungesättigte Fette handelt. Mehrfach ungesättigte Fettsäuren senken die Mortalität dabei stärker als einfach ungesättigte Fettsäuren. Ersetzt man allerdings Kohlenhydrate durch gesättigte Fette, führt das zu einer leichten Steigerung der Mortalität, während ein Ersatz durch Trans-Fette zu einer starken Steigerung der Mortalität führt.
12. NAHRUNGSPROTEINE, BEGINN DER PROTEINVERDAUUNG IM MAGEN
Wie decken wir
unseren Bedarf an Nahrungsmittelprotein? Nahrungsmittel tierischen Ursprungs
enthalten die "richtige" Mischung an Aminosäuren. Neben ethischen und Klimawandelbedenken
hat diese Lösung jedoch auch einen ernährungsphysiologischen Haken: Fleisch,
Milchprodukte, Eier enthalten, wie wir gerade gesehen haben, die
"falschen", nämlich ganz überwiegend gesättigte, Fettsäuren, zudem
beträchtliche Mengen an Cholesterol.
Pflanzliche
Nahrungsmittel enthalten die "richtigen" Fette, dafür jeweils eine
"falsche" Mischung an Aminosäuren. Den relativen Mangel an Lysin in Getreiden
und Reis haben wir uns bereits bewusst gemacht; bei Mais ist zudem Tryptophan
limitierend, Hülsenfrüchte weisen einen reduzierten Gehalt an schwefelhaltigen
Aminosäuren auf, etc.
Die
traditionelle, "altsteinzeitliche" Lösung dieses Problems besteht
darin, wenig Fleisch und viel Pflanzliches zu essen. Das ist auch für Kinder
mit ihrem hohen Bedarf an ausgewogenem Protein, Eisen, B12 und Ca2+ zu empfehlen.
Möchte man auf tierische
Nahrungsmittel ganz verzichten, muss man auf eine Mischung der pflanzlichen
Proteinquellen achten. Relativ viel Protein enthalten Hülsenfrüchte
(Sojabohnen, Bohnen, Erbsen, Linsen, Kichererbsen, Erdnüsse), Ölsaaten
(Kürbiskerne, Sonnenblumenkerne) und Nüsse (Mandeln, Walnüsse, Haselnüsse). Als
Basis eignet sich wegen ihrer relativ besten Aminosäureverteilung die
Sojabohne/Edamame/Tofu, doch muss ihr niedriger Gehalt an schwefelhaltigen
Aminosäuren (Methionin, Cystein) aus anderen pflanzlichen Quellen wie Getreide
kompensiert werden. Die Strategie ist also denkbar einfach: kombiniere
Hülsenfrüchte mit Getreide. Reis mit Bohnen hat sich auch in weniger begüterten
Weltgegenden als Basisgericht bewährt.
Mütter, die sich vegan oder vegetarisch ernähren und das auch für ihre Babys anstreben, sollten ihre Säuglinge stillen. Damit bekommen diese automatisch ausreichend Protein in der richtigen Aminosäurezusammensetzung (und in der Regel ausreichend Docosahexaensäure). Falls das nicht möglich oder erwünscht ist, bleibt nur die Möglichkeit einer Säuglingsnahrung auf Sojabasis. Diese ist angereichert mit Methionin; ebenso ist sie angereichert mit Eisen und Zink, da sie Phytinsäure enthält, die, wie erwähnt, Eisen und Zink bindet. Ein kleiner Bereich der Unsicherheit ergibt sich daraus, dass in dieser Säuglingsnahrung Soja-Isoflavone (Genistein, Daidzein und Glycitein) enthalten sind, die als Phytoöstrogene wirken. Mögliche negative Folgen im späteren Leben dieser Kinder können noch nicht sicher ausgeschlossen werden.
In psychologischen Experimenten
über zwischenmenschliche Verhandlungen in Rollenspielen wurde gezeigt, dass Toleranz/Strafbereitschaft
durch die Zusammensetzung einer vorangegangenen Mahlzeit beeinflusst werden,
insbesondere durch deren Proteingehalt. Tyrosin ist die Vorstufe von Dopamin
und Noradrenalin, Tryptophan die Vorstufe von Serotonin. Ein sehr
hypothetischer, wohl übersimplifizierter Erklärungsversuch für die beobachtete
"Beeinflussung des freien Willens" wäre also das Angebot dieser
Neurotransmittervorstufen im Blut. Schwierige Probleme bei einem gemeinsamen
Essen zu lösen ist jedenfalls ein ratsamer Ansatz.
Ein aktueller, von Geschäftemachern geschickt
lancierter Hype baut darauf auf, den Menschen die Sorge einzureden, zuwenig
Protein zu sich zu nehmen. Ziel ist der Verkauf von teuren,
proteinangereicherten Produkten, Proteinshakes oder -riegeln. Das ist gänzlich
unnotwendig, sogar kontraproduktiv. Die empfohlene Tagesmenge für Erwachsene
ist 0,8 g pro kg Körpergewicht, d. h. für unsere 70 kg-Einheitsperson 56 g
Protein pro Tag. Diese recommended daily
allowance berücksichtigt die Normalverteilung an Proteinbedarf in der
Population: Man ermittelt den Mittelwert und addiert dazu die zweifache
Standardabweichung. Mit diesem Vorgehen nehmen bei 0,8 g/kg Körpergewicht 97,5%
der Menschen MEHR Protein zu sich, als sie brauchen. Bleiben 2,5% –
hauptsächlich muskulöse, schwer gebaute Männer –, die noch ein wenig mehr, aber
nicht viel mehr brauchen. Diese Menge an Protein nimmt man mit den allermeisten
Formen der Ernährung locker auf, ohne sich jemals Gedanken darüber machen zu
müssen. Wenn man mit einer einigermaßen variierten Kost ausreichend Kalorien zu
sich nimmt, sodass das Gewicht stabil bleibt, nimmt man automatisch auch ausreichend
Protein auf, unabhängig, ob man Veganer, Flexitarier oder sonstwas ist.
Tatsächlich nehmen die meisten von uns das Doppelte der empfohlenen Tagesmenge
auf. Ein Überschuss an Proteinzufuhr ist nicht speicherbar und führt nicht zu
mehr Muskel, sondern wird zu Fett umgewandelt. Zusätzlich belastet der dabei
erhöhte Anfall von Stickstoff die Niere. Der Proteinbedarf pro kg erhöht sich
allerdings bei bestimmten Gruppen:
·
bei
Schwangeren und stillenden Müttern, sowie bei Kindern im Wachstum
·
bei alten
Menschen, die das Protein weniger effizient verwerten. Zur Erhaltung ihrer Muskel- und Knochenmasse benötigen
sie einerseits regelmäßiges Resistenztraining, andererseits 1,2-1,5 g
Protein/kg Körpergewicht.
·
bei
Menschen, die gerade ein Resistenztrainingsprogramm mit dem Ziel von
Muskelaufbau durchlaufen (und das auch tatsächlich systematisch tun, nicht nur
wollen).
·
bei
Menschen, die GLP‑1-Analoga zur Gewichtsreduktion anwenden, um den
Prozentsatz verlorener Muskelmasse zu limitieren, wiederum in Verbindung mit
Resistenztraining.
*
Die Verdauung der Nahrungsproteine
beginnt in der "Mischmaschine" Magen durch das Zusammenwirken
von Säure und Pepsinogen. Für die Verdauung von Kohlenhydraten und Fetten
spielt der Magen nur eine geringe Rolle.
Proteine sind relativ schwer
zu "knacken". Die Faltung schützt die meisten Proteine vor
dem Angriff durch Proteasen. Der erste Schnitt ist der schwierigste.
Ist der einmal gemacht, verlieren die entstehenden Bruchstücke einen
Teil dieser Faltung und können leichter weiter abgebaut werden. Sehr
hohe Protonendichte (der pH im Magen erreicht Werte um 1) denaturiert
(entfaltet) Teile der Proteine und ermöglicht so die ersten Schnitte.
Proteasen werden danach eingeteilt, welche chemische Gruppe sie verwenden,
um eine Peptidbindung aufzubrechen: so gibt es Serin-, Cystein-, Aspartat-
und Metalloproteasen. Die Aktivität von Endopeptidasen, die Peptidbindungen
im Inneren einer Aminosäurekette aufbrechen, ist von der Aminosäuresequenz
des Substrats abhängig. Pepsin ist eine Aspartat-Protease, die neben
hydrophoben Aminosäuren schneidet; besonders effizient nach Phenylalanin.
Um Proteine effizient abzubauen, wird daher eine Reihe von Proteasen
benötigt, die an verschiedenen Stellen schneiden.
Die Bestandteile des Magensekrets
werden von den zahlreichen Drüsen der Magenschleimhaut sezerniert. In
die Epithelschicht sind unterschiedliche Zelltypen integriert. Nebenzellen
produzieren schützenden Schleim, Hauptzellen Pepsinogen und Magenlipase, (neuro)endokrine
Zellen Signalmoleküle. Dazu gehören z. B. im Antrum G-Zellen, die das
Signalpeptid Gastrin produzieren, und D-Zellen, die Somatostatin sezernieren,
das der Gastrinproduktion entgegenwirkt. Stammzellen und deren noch
undifferenzierte Nachkommen, transit
amplifying cells, sorgen für Zellnachschub. Die Salzsäure wird von
Beleg- oder Parietalzellen gebildet.
Belegzellen können auf
Stimulation ihre apikale Membranoberfläche durch Ausbildung intrazellulärer
Kanal-Labyrinthe mit Mikrovilli um den Faktor 50-100 vergrößern. Basis
der Salzsäureproduktion ist eine H+/K+-ATPase,
die Protonen im Austausch gegen K+ in das Lumen pumpt. K+
strömt durch K-Kanäle wieder ins Lumen zurück und Chlorid-Ionen folgen
durch Chloridkanäle. Die Protonen werden durch Aufnahme von CO2
und H2O in die Parietalzelle generiert, die durch Carboanhydrase
zu H+ und HCO3- umgesetzt werden. HCO3- verlässt die Zelle basolateral
durch Austausch gegen Chlorid. Netto resultiert die Abgabe von HCl in
das Magenlumen. Diese Sekretion wird durch Vagus-Neuronen über Acetylcholin,
sowie über Gastrin aus G-Zellen im Antrum angekurbelt. Diese beiden
Stimuli wirken einerseits direkt an den Parietalzellen, andererseits
indirekt, indem sie die Ausschüttung von Histamin aus neuroendokrinen
Zellen unter dem Epithel, den ECL-(enterochromaffin-like)
Zellen, bewirken. Parietalzellen rezipieren das Histamin über H2-Rezeptoren.
Pharmakologische Querverstrebung: Protonenpumpenhemmer wie Omeprazol binden kovalent
an Cysteine auf der zytosolischen Seite der Pumpe und inaktivieren diese irreversibel.
Die Wirkung hält länger als einen Tag an, bis eine ähnliche Zahl neuer
Pumpenproteine nachproduziert wird. Kompetitive H2-Rezeptorantagonisten
wie Ranitidin bremsen die Säureproduktion ebenfalls, wurden aber durch
die Protonenpumpenhemmer weitgehend verdrängt.
Pepsinogen ist das Produkt
der Hauptzellen. Es wird über eine Reihe von neuronalen und Hormonstimuli freigesetzt, von denen Acetylcholin aus Vagus-Neuronen der wichtigste
ist. Pepsinogen benötigt Säure. Bei niedrigem pH spaltet es sich spontan
in ein N-terminales Peptid und das aktive Pepsin. Dieser Prozess wird
erst unter pH 3 effizient und dadurch positiv rückgekoppelt, dass
dann auch Pepsin selbst Pepsinogen spaltet. Pepsin ist eine Endopeptidase
mit pH-Optimum zwischen 1,8 und 3,5; darüber ist Pepsin inaktiv. Das
Produkt der Pepsinspaltung sind große Peptide, sogenannte Peptone, die
ihrerseits G-Zellen im Magenantrum zur Sekretion von Gastrin und I-Zellen
in der Duodenalschleimhaut zur Sekretion von Cholezystokinin anregen.
Um die Aufgaben der Magensäure
zusammenzufassen: Säure erleichtert das Spalten von Peptidbindungen;
bei sehr niedrigem pH beschleunigt sich bereits die spontane Hydrolyse
von Peptidbindungen wesentlich. Säure bewirkt auch eine Denaturierung
der Proteine, die diese erst für Proteasen angreifbar macht. Schließlich
tötet die Magensäure den Großteil aller Bakterien ab und schützt uns
damit vor Infektionen. Diesen Vorteilen steht der Nachteil gegenüber,
dass Salzsäure (HCl) natürlich dieselben Wirkungen auf die Zellen der
Magenschleimhaut hat. Die einzige Möglichkeit, die Epithelschicht vor
schweren Gewebsschäden zu bewahren, ist der Schutz durch einen dauernd
erneuerten Schleimfilm mit neutralem Milieu.
Schleimproduzierende Zellen
sitzen an der Oberfläche des Magens, in den Eingängen der Magendrüsen
und in den Drüsen selbst. Schleim besteht aus Tetrameren des Glycoproteins
Mucin, die lange sulfatierte Kohlenhydratketten binden, die einen großen
Wassermantel tragen. Dieser dauernd nachproduzierte Schleimteppich wird
durch HCO3- alkalisiert und bildet damit eine
neutralisierende Diffusionsbarriere für das Magenepithel. Der fließende
Teppich wird von unten ständig nachproduziert und von oben ständig verdaut.
Von den Schleimzellen produziertes HCO3- bleibt
in den Maschen gefangen; aktives Pepsin wird vom Maschenwerk draußen
gehalten. Die wenigen eindringenden Pepsinmoleküle werden durch den
höheren pH inaktiviert. Die Produktion von HCO3-
ist abhängig von lokal produziertem Prostaglandin E (PGE), das über
die Cyclooxygenase 1 (COX1) hergestellt wird.
Pharmakologische Querverstrebung: Wollen wir Medikamente oral verabreichen,
stellt die Magensäure oft eine Herausforderung für die Galenik dar. Manche
Medikamente würden durch die Magensäure inaktiviert werden, andere die
Magenschleimhaut schädigen. Ein Beispiel ist der Protonenpumpenhemmer Omeprazol
selbst, der im Magen vor Säure geschützt werden muss. Eine Möglichkeit ist, solche
Tabletten mit einer magensaftresistenten Schicht zu überziehen. Allerdings
führt das oft dazu, dass die Tablette lange im Magen liegen bleibt. Der Magen
funktioniert ja als "Mischmaschine", die den Zweck hat, proteinreiche
Nahrung lange im sauren Milieu zu halten, wobei die Peristaltik beträchtliche
Drücke erzeugt. Damit keine zu großen Mengen in das Duodenum hinausgedrückt
werden, kontrahiert sich der Pylorus, sodass nur Brei und Partikel kleiner als
2 mm Durchmesser hindurch gelangen. Partikel, die größer sind, bleiben
paradoxerweise im Magen liegen, bis dieser entleert ist; erst dann erschlafft
der Pylorus und entlässt die letzten größeren Partikel. Eine so genannte
monolithische magensaftresistente Tablette bleibt also, besonders, wenn jemand
regelmäßig etwas "dazu isst", über viele Stunden im Magen und wird
möglicherweise erst in der Nacht (oder drei Tabletten gleichzeitig, bei 3x1!)
entlassen und damit wirksam. Gelöst werden kann das Problem durch so genannte
MUPS (multi-unit pellet system), bei
denen separat magensaftresistente Pellets kleiner als 2 mm Durchmesser zu einer
Tablette zusammengefasst werden. Die Tablette zerfällt im Magen, die Pellets
können den Pylorus passieren. Teuer!
13. GASTRITIS UND ULCUS VENTRICULI / DUODENI
Eine Verminderung des Schutzwalles
mit oberflächlichen Erosionen finden wir bei der Gastritis. Beim Ulcus
ist die Schleimhaut lokal völlig durchbrochen. Dieser Substanzdefekt
in der Magenwand ist im sauer-proteolytischen Dauerbeschuss extrem schwer
zu schließen. Bis zur Entwicklung der Protonenpumpenhemmer und der Erkenntnis,
das die behandelbare Infektion mit Helicobacter
eine pathogenetische Rolle spielt, war es vielfach der einzige Ausweg,
den Großteil des Magens operativ zu entfernen.
Helicobacter pylori ist säureresistent und kolonisiert besonders die
Schleimhaut des Antrums, da im Fundus die Parietalzellen am dichtesten sind und
damit die höchste Säurekonzentration herrscht. Das Bakterium bewegt sich im
Säuregradienten chemotaktisch Richtung neutraleres Milieu, daut sich durch
Proteasen in die Schleimschicht ein und dockt an Magenepithelzellen an. Es
stellt seinen eigenen HCO3−-Puffer durch Spaltung
von Harnstoff mittels Urease her. Das dabei entstehende NH4+ ist, wie die Proteasen, toxisch für die danebenliegende Schleimhautzelle. Nur
ein Teil der H. Pylori-Stämme ist pathogen. Diese enthalten einen
Genomabschnitt, der als cag-pathogenicity
island (cag- cytotoxin-associated gene) bezeichnet wird. Die dort kodierten Gene
befähigen das Bakterium, Peptidoglykan ins Innere der Schleimhautzelle zu
injizieren, das über NOD-like receptors erkannt wird und eine Entzündung
auslöst, welche die Schleimhautwand auflockert. Das Eindringen in die
Schleimhautwand verschafft Helicobacter eine höhere
Überlebenswahrscheinlichkeit. Durch die Veränderungen wird auch mehr Gastrin
und damit insgesamt mehr Säure gebildet. Obwohl nur ein Teil der Träger
Beschwerden bekommt, ist H. pylori für den Großteil der Magen- und Zwölffingerdarmgeschwüre verantwortlich; an
zweiter Stelle liegen die NSAIDs, die weniger als ein Fünftel der Geschwüre
auslösen. Zehn bis zwanzig Prozent der Infizierten entwickeln im Lauf ihres Lebens
ein Ulcus. Die Infektion kann durch Nachweis der Spaltung von 13C-
oder 14C-radioaktiv markiertem Harnstoff im Atemtest nachgewiesen
werden.
Pharmakologische Querverstrebung: Zwei extrem häufig zur Entzündungshemmung eingesetzte
Medikamentfamilien, Glucocorticoide und nicht-steroidale Entzündungshemmer
(NSAIDs), hemmen über unterschiedliche Mechanismen die Produktion
von PGE. Da dies auch in der Magen- und Darmschleimhaut geschieht, macht
der dadurch verminderte Schleimhautschutz Magenulcera und Blutungen
zu gefürchteten Nebenwirkungen längerdauernder anti-entzündlicher Therapien.
Um diesem Effekt entgegenzuwirken, werden Protonenpumpenhemmer als "Magenschutz"
eingesetzt. Leider nützt
dieser "Schutz" nur im Magen, nicht aber in den Darmabschnitten
hinter dem Magen, während die NSAIDs-Schädigung sich dort natürlich ebenso
auswirkt. Weiters wurden, um die in allen Zellen konstitutiv exprimierte
COX‑1 weniger zu beeinträchtigen, COX-2-Hemmer entwickelt, um
präferentiell diese in Makrophagen und anderen Entzündungszellen induzierbare
Isoform zu hemmen. Diese Entwicklung war nur teilweise erfolgreich,
da COX‑2-Hemmer zwar weniger magenbelastend sind, doch eine verstärkte
Neigung zu atherothrombotischen Ereignissen mit sich bringen.
14. PROTEINVERDAUUNG UND AUFNAHME
Aus dem Magen werden kleine
Portionen Chymus (Speisebrei) vom Pylorus (Pförtner) in den Zwölffingerdarm
entlassen. Dieses Gemisch ist sauer und enthält das Resultat der Pepsinwirkung,
Peptone. Der niedrige pH stimuliert S-Zellen der Duodenalschleimhaut
zur Freisetzung von Sekretin, das Pankreasgangzellen zur Sekretion von
HCO3‑ stimuliert. Peptone
stimulieren I-Zellen im Duodenalepithel zur Freisetzung von Cholecystokinin
(CCK). CCK stimuliert den Gallefluss durch Kontraktion der Gallenblase
und regt die Bauchspeicheldrüse zur Freisetzung ihres Enzymcocktails
an. Gemeinsam führen Sekretin und CKK also zur Freisetzung der Pankreas-Verdauungsenzyme
in einem reichlichen Volumen alkalischen Sekrets, das den Speisebrei
neutralisiert.
Im Duodenum wir die Proteinverdauung
durch eine Reihe pankreatischer Enzyme fortgesetzt, die ihr pH-Optimum
im leicht alkalischen Bereich haben. Diese werden ebenfalls als inaktive
Vorstufen sezerniert: Trypsinogen, Chymotrypsinogen, Proelastase, Proprotease
E sowie die Procarboxypeptidasen A und B. Trypsinogen wird durch das
Bürstensaum-Enzym Enteropeptidase (Enterokinase) durch Abspaltung eines
Peptids zu Trypsin aktiviert. Trypsin aktiviert sowohl weitere Trypsinogenmoleküle,
als auch die anderen Proteasen und Peptidasen. Trypsin ist eine Serinprotease,
die Aminosäureketten nach den basischen Aminosäuren Lysin und Arginin
spaltet. Das verwandte Chymotrypsin, ebenfalls eine Endopeptidase, spaltet
nach Phenylalanin, Tyrosin, Tryptophan und Methionin. Die Carboxypeptidasen
sind Exopeptidasen, knabbern also die Aminosäuren einzeln vom C-Terminus
ab. Das Resultat dieses Verdauungsvorgangs sind einzelne Aminosäuren
(etwa 30%) und Peptide aus wenigen Aminosäuren (70%).
Den Rest besorgen die Enterozyten
des Dünndarms in zwei Stufen: durch zahlreiche Bürstensaum-Peptidasen
sowie zahlreiche cytosolische Peptidasen. Bürstensaum-Exopeptidasen
verkleinern die bestehenden Peptide zunächst weiter, und Transporter
für Tetra-, Tri- und Dipeptide (PepT1) sowie für einzelne Aminosäuren
sorgen für deren Aufnahme in Enterozyten, als Kotransport mit Protonen
oder Na+. Tetra-, Tri- und Dipeptide werden im Cytosol des
Enterozyten in einzelne Aminosäuren zerlegt und diese basolateral ins
Pfortaderblut ausgeschleust.
Durch die bisher beschriebenen Mechanismen werden nur Aminosäuren aufgenommen. Vergessen wir dabei nicht, dass in geringem Ausmaß aber auch größere Peptide und komplexe Proteine durch das Epithel geschleust werden. Dafür sind besonders die M-Zellen im Epithel über den Peyer-Plaques verantwortlich (siehe Skript Immunologie). Dies hat den Sinn, das Immunsystem über die Antigensituation im Darmlumen zu informieren.
15. PANKREATITIS
Mehr noch als das Magensekret
stellt das Pankreassekret eine biochemische Bombe dar. Diese ist gewöhnlich
sorgfältig gesichert. Proteasen werden als inaktive Vorstufen produziert.
Sie werden in sekretorischen Granula von anderen Zellbestandteilen ferngehalten.
In diese Granula werden Enzymhemmer mitverpackt: der Hauptschalter Trypsinogen
wird zusätzlich durch einen pankreatischen Trypsininhibitor gesichert,
der bis zu 10% "versehentlich" aktiviertes Trypsin abfangen
kann. Der Inhalt der Granula wird auf einen niedrigeren pH-Wert gebracht,
bei dem die Enzyme nicht aktiv sind.
Trotzdem kommt es manchmal
zu einer Aktivierungskaskade noch im Pankreas, das sich dann prompt
selbst zu verdauen beginnt. Die häufigsten Auslöser sind Gallensteine,
die in der Papilla Vateri
stecken bleiben, und kräftiger Alkoholkonsum. Die Kausalkette von diesen
Auslösern zur akuten Pankreatitis ist noch unzureichend geklärt. Bei
Gallenkonkrementen schädigen die zurückgestauten Gallenbestandteile
die Pankreas-Zellmembranen, sodass diese permeabler werden. Es ist auch
möglich, dass durch eine Ventilfunktion etwas Duodenalinhalt mit aktiviertem
Trypsin in das pankreatische System zurück fließt. Auch bei Alkoholkonsum
kann die Stimulierung zu einem Stau führen, bei der die bereits in der
aktiven Form sezernierte Lipase die Membranen schädigt.
Einige Polymorphismen erleichtern
die Entstehung einer Pankreatitis, die dadurch auch familiär auftreten
kann. Das Trypsinogen im Pankreassekret ist das Produkt mehrerer ähnlicher
Gene. Die Varianten können durch unterschiedliche Ladung elektrophoretisch
unterschieden werden. Die Mutation Arg122His in der Hauptvariante, dem
sogenannten kationischen Trypsinogen (Gen: PRSS1 für Protease, Serine, 1), führt dazu, dass einmal aktiviertes Trypsin
länger aktiv bleibt, da es durch ein anderes Trypsinmolekül nicht mehr
geschnitten werden kann. Ein solches Allel genügt; diese Form der Pankreatitis-Prädisposition
verhält sich daher autosomal dominant. Mutationen im Trypsininhibitor
(Gen: SPINK1 für Serine Protease
Inhibitor, Kazal-type 1) verhindern das Auffangen von autolytisch
aktivierten Trypsinogenmolekülen und verhalten sich autosomal rezessiv.
Akute Pankreatitis kann
lebensbedrohlich sein. Die Aktivierung der Selbstverdauung führt zur
Freisetzung des Enzymcocktails und zur sekundären Aktivierung von Entzündungszellen
und Endothel im ganzen Körper. Folgen können disseminierte intravaskuläre
Koagulation, Hämolyse, Schock, Nierenversagen und acute respiratory distress syndrome (ARDS)
sein.
Eine chronische Pankreatitis kann mit der Zeit zu
einer exokrinen Pankreasinsuffizienz führen, die eine Malnutrition zur Folge
hat und Symptome wie Bauchschmerzen, Diarrhoe, Fettstühle. Unbehandelt kann
sich starke Osteoporose entwickeln. Die Elastase des Pankreassekrets ist sehr
stabil gegenüber den anderen Proteasen; ein beträchtlicher Teil davon wird im
Stuhl ausgeschieden und kann zur Diagnose gemessen werden. Eine Reduktion der
ausgeschiedenen Elastasemenge spricht für eine exokrine Pankreasinsuffizienz. In
diesem Fall müssen die Pankreasenzyme oral substituiert werden.
16. ZYSTISCHE FIBROSE (MUKOVISZIDOSE)
Zystische Fibrose ist die
häufigste lebensbedrohliche genetische Erkrankung von Menschen Europäischen
Ursprungs; die Häufigkeit der autosomal rezessiven Erkrankung beträgt
etwa 1 in 2000. Der Name "zystische Fibrose" bezieht sich
auf die typische Zerstörung des Pankreasgewebes, die durch den Mangel
an Verdauungsenzymen zu schweren Ernährungsstörungen und frühem Tod
der Patienten führte. Seit die Enzyme oral substituiert werden können,
stellen Lungenerkrankungen das zentrale Problem dar. Eine zähe Schleimschicht
behindert den Transport durch das Flimmerepithel und stellt einen idealen
Nährboden für Infektionen mit Pseudomonas aeruginosa und Staphylococcus aureus dar.
Zystische Fibrose wird
durch Mutationen im CF-Gen verursacht, das den cystic fibrosis transmembrane conductance regulator (CFTR) kodiert.
CFTR ist notwendig, um das HCO3‑-reiche
Pankreassekret zu erzeugen. Dabei ist CFTR eigentlich ein Cl--Transporter auf der apikalen
Membran des Pankreasgangepithels, doch wird das zunächst ausgeschleuste
Cl‑ gebraucht,
um es wieder gegen das intrazellulär befindliche HCO3‑ einzutauschen. CFTR
besteht aus zwei Transmembrandomänen mit benachbarten ATP-Bindungsstellen,
die eine intrazelluläre Domäne mit zahlreichen Phosphorylierungsstellen
für PKA und PKC umschließen. Die Aktivierung von PKA erfolgt via cAMP
durch Sekretin, jene von PKC über parasympathisches Acetylcholin. In
Summe aktivieren Verdauungsstimuli also die Produktion von HCO3‑reichem Sekret über die Phosphorylierung von CFTR.
Eine Mutation, ΔF508, ist für etwa zwei Drittel aller CF-Fälle verantwortlich. Die Deletion
von Phenylalanin 508 führt zu einer Fehlfaltung des Proteins, sodass
es rasch degradiert wird und gar nicht die apikale Membran erreicht.
Zahlreiche weitere Mutationen, in Summe mehr als 1000, wurden in jeweils nur einem geringen Anteil
der Patienten gefunden. Manche von diesen haben einen totalen, andere
nur einen teilweisen Funktionsausfall zur Folge. CFTR wird für die Funktion
vieler Epitheltypen benötigt. Der Ausfall von CFTR kann daher ein breites
Spektrum von weiteren Symptomen mit sich bringen, z. B. Mekoniumileus
bei Neugeborenen, Nasennebenhöhlenentzündungen, Infertilität, sekundären
Diabetes oder biliäre Zirrhose.
Pharmakologische Querverstrebung: Bei Patienten mit einer homozygoten ΔF508-Mutation kann die Kombination Elexacaftor/Tezacaftor/Ivacaftor
(Tablette, Trikafta®) die Symptome verbessern: Elexacaftor und
Tezacaftor haben Chaperonwirkung, sodass ein kleiner Anteil der CFTR-Proteine
korrekt faltet, die Membran erreicht und durch Ivacaftor eher offen bleibt.
EINIGE NICHT-INFEKTIÖSE DARMERKRANKUNGEN
Zöliakie wird auch als
Gluten-sensitive Enteropathie oder, heute selten, als Sprue bezeichnet.
Die Darmerkrankung kann in genetisch prädisponierten Individuen durch
bestimmte Proteine aus Weizen, Roggen oder Gerste ausgelöst werden.
Hafer wird von den meisten Patienten toleriert, ist aber häufig mit
anderen Getreiden kontaminiert. Mais und Reis sind unproblematisch.
Mit einer Prävalenz von 0,5 bis 1% ist die Erkrankung bei Menschen mit Europäischem genetischen Hintergrund häufig; sie ist weniger häufig bei anderen Populationen. Erinnern wir
uns: Getreide ist erst seit etwa 10.000 Jahren ein wesentlicher Teil
der menschlichen Ernährung.
Die Nomenklatur von Getreideproteinen
ist für Nichtexperten ziemlich verwirrend. Um die Keimung zu ermöglichen,
enthält ein Getreidekorn Kohlenhydrate (in Form von Stärke) und Aminosäuren
(in Form von Speicherproteinen). Stärke sowie ein Teil der Proteine
sind wasserlöslich. Das Gemisch der nicht-wasserlöslichen Proteinfraktion
wird als Gluten bezeichnet, abgeleitet vom lateinischen Wort gluten, für Klebstoff. Die in Alkohol lösliche
Subfraktion dieser Glutenproteine
werden Prolamine genannt, weil sie viele Proline und Glutamine enthalten.
Prolamine haben in den einzelnen Getreidesorten unterschiedliche Namen:
in Weizen werden sie Gliadine genannt, in Gerste, Hordeine und in Roggen,
Secaline. Weizen ist die in unserer Ernährung vorherrschende Getreidesorte;
daher spielen Gliadine, von denen es α/β-, γ- und ω‑Formen
gibt, die größte Rolle.
Da unsere Verdauungsproteasen
Peptidbindungen neben Glutamin und Prolin nur unzureichend schneiden,
sind Gliadine widerstandsfähig gegen enzymatische Verdauung im Darm.
Der Verdauungsprozess resultiert daher in typischen Peptidfragmenten,
die z. B. die Größe von 10-35 Aminosäuren haben. In prädisponierten
Personen lösen diese Peptidfragmente auf zwei Arten Immunreaktionen
aus.
Gliadinpeptide haben direkte
Wirkungen auf Enterozyten, über erst zum Teil aufgeklärte Mechanismen.
Manche Gliadin-Peptide haben eine gewisse Affinität zum Chemokinrezeptor
CXCR3. Aktivierung von CXCR3 führt zur Sekretion von Zonulin (Prä-Haptoglobin 2),
einem Protein, das die tight junctions
durchlässiger macht, sodass mehr Gliadinpeptide die Epithelschranke
überwinden können. Zudem können so durch Gliadinpeptide CXCR3-exprimierende
Lymphozyten angezogen werden.
Gliadinpeptide stimulieren
Enterozyten auch zur Sekretion von IL‑15 und zur Expression von
MICA, einem MHC-I-verwandten Transmembranprotein. IL‑15 stimuliert
intraepitheliale Lymphozyten zur Expression von MICA-Rezeptor NKG2D.
Über diesen nicht-adaptiven Mechanismus töten die Lymphozyten Epithelzellen,
sodass die Darmwand noch durchlässiger wird. Überdies führt der Mechanismus
mit der Zeit zu einer Reduktion der Zahl an Enterozyten und damit zu
einer Schleimhautatrophie.
Ein zweiter Mechanismus
beruht darauf, dass das ubiquitär exprimierte Enzym Gewebstransglutaminase
(tissue transglutaminase, tTG) Glutamin
zu Glutaminsäure umwandelt. Die desaminierten, nun negativ geladenen
Gliadinfragmente werden präferentiell in den Peptidbindungsspalt bestimmter
MHC-II-Moleküle eingebaut und so präsentiert. Die dazu geeigneten MHC-II-Moleküle
sind Varianten von HLA-DQ2.5 (Heterodimere, die
von DQA1*05:01 und DQB1*02:01-Allelen stammen) oder HLA-DQ8 (Heterodimere von
DQA1*03:01 und DQB1*03:02-Allelen). Diese Allele sind sehr häufig; ihr
Vorhandensein in einem Individuum bedeutet nicht, dass eine Zöliakie
zu erwarten ist, doch sind andere DQ-Typen offensichtlich nicht in der
Lage, dieselbe Funktion auszuüben. Antigen-präsentierende Zellen (APC)
präsentieren die desaminierten Gliadinpeptide naiven CD4-positiven T-Zellen
und aktivieren diese. Die T-Zellen differenzieren hauptsächlich zu TH1-Zellen,
die IFNγ produzieren und Makrophagen aktivieren. Damit lösen sie
eine chronische Entzündung der Darmwand aus. Da diese Mechanismen eine
zelluläre Immunreaktion darstellen, handelt es sich um eine Form der
Typ IV-Überempfindlichkeitsreaktion.
Gemeinsam führen die beiden
Mechanismen zu einem Verlust reifer Enterozyten, der durch verstärkte
Proliferation der transit amplifying-Vorläufer
in den Krypten nur unzureichend wettgemacht werden kann. Die diagnostische
Dünndarmbiopsie zeigt demgemäß Villusatrophie, Kryptenhyperplasie und
eine große Zahl an intraepithelialen Lymphozyten.
Vor der Biopsie
empfehlen sich Laboruntersuchungen auf IgA-Antikörper gegen
Gewebstransglutaminase (tTG). Diese diagnostisch hilfreichen Antikörper sind
nach gegenwärtigem Wissen eine Nebenerscheinung, keine Krankheitsauslöser.
Gewöhnlich kommen keine tTG-spezifischen T-Helferzellen vor- sie sollten auch
nicht vorkommen, da tTG ja ein normales Selbst-Molekül ist. Die entsprechenden
B-Zellen bekommen ihre Hilfe von Gliadin-spezifischen Helferzellen, wenn
B-Zellen mit einem tTG-spezifischen B-Zell-Rezeptor einen Komplex aus tTG und
Gliadin internalisieren und in der Folge selbst ein Gliadinpeptid auf ihren
DQ-Molekülen präsentieren. Gliadin-spezifische T-Zellen geben also die Hilfe
zur Produktion eines tTG-spezifischen Autoantikörpers. Einen analogen
Mechanismus haben wir bereits bei den Konjugatimpfstoffen kennengelernt.
Zöliakie kann in jedem
Alter und in sehr verschiedener Intensität beginnen, und zeigt oft uncharakteristische
Symptome. Das Defizit an funktionellen Enterozyten kann schleichend
zu Mangelernährung führen, z. B. in Hinsicht auf Eisen, Ca2+ oder einzelne
Vitamine. In voller Ausprägung führt Zöliakie zu chronischen Durchfällen
mit starker enteraler Gasbildung, Gedeihstörungen und Gewichtsverlust.
Die einzige wirksame Therapie besteht in einer
lebenslangen glutenfreie Ernährung.
Manche Menschen haben weder eine Zöliakie noch
eine klassische Allergie gegen Weizenbestandteile, vertragen aber
weizenbasierte Nahrungsmittel trotzdem nicht gut. In solchen Fällen werden ATI
(Amylase-Trypsin-Inhibitoren, Glutenproteinbestandteile) und FODMAPS (fermentable oligosaccharides, disaccharides,
monosaccharides, and polyols) diskutiert. Die Idee ist, dass FODMAPS durch
manche Individuen schlechter verdaut werden können und/oder ATI-Bestandteile
des Glutens den Verdauungsprozess verzögern. In beiden Fällen würde das zur
vermehrten Fütterung von Darmbakterien mit entsprechenden intestinalen
Symptomen führen.
Zu FODMAPS gehören hoher Laktose- und Fructosegehalt aus den vorher diskutierten Gründen, aber auch Sorbitol, Fructane und Galaktooligosaccharide. Relativ hoher Gehalt an FODMAPS findet sich daher in zahlreichen alltäglichen Lebensmitteln, welche die allermeisten von uns problemlos konsumieren, z. B. in Milchprodukten – Milch, Joghurt, Eis, Weichkäse – , Getreide-basierten Produkten – Nudeln und Brot –, aber auch in vielen Leguminosen – Bohnen und Linsen –, manchen Gemüsen – Lauch, Zwiebel, Knoblauch, Karfiol, Spargel –, manchen Früchten – Äpfel, Birnen, Kirschen, Pfirsiche, Pflaumen –, in Honig (Fructose) sowie in manchen Nüssen – Pistazien und Cashews. Ein FODMAP-arme Diät ist daher sehr restriktiv, vom Ernährungsstandpunkt aus gesehen problematisch und eher ultima ratio. Zu den Nahrungsmittlen mit niedrigem FODMAP-Gehalt gehören Eier, Hartkäse, nicht-prozessiertes Fleisch, Fisch, Tofu, Reis, andere Gemüse – Kartoffeln, Karotten, Auberginen, Paprika, Salatgurken, Salat – andere Früchte – Zitrusfrüchte, Kiwis, Zuckermelonen –, andere Nüsse – Erdnüsse, Walnüsse. Wenn eine FODMAP-arme Diät nach 2-4 Wochen keine Symptombesserung bringt, kann man darauf verzichten, dann waren nicht FODMAPS die Ursache. Bringt die Diät eine merkliche Besserung, sollte man nicht dabei stehen bleiben, sondern langsam andere Lebenmittel wieder hinzunehmen, bis man eine individuell angepasste, verträgliche Nahrungsmittelzusammensetzung erreicht.
18. INFLAMMATORY BOWEL DISEASE
Eine ungerechtfertigte
oder überschießende Aktivierung des Immunsystems in der Darmwand ist
das gemeinsame Merkmal zweier Erkrankungen, Morbus
Crohn und Colitis ulcerosa, die unter der Bezeichnung inflammatory bowel disease (IBD) zusammengefasst
werden (die direkte deutsche Übersetzung, "entzündliche Darmerkrankung",
hat nicht dieselbe begriffliche Bedeutung). Bei vielen Ähnlichkeiten
unterscheiden sich die beiden Erkrankungen in der Lokalisation und in
der Eindringtiefe des entzündlichen Geschehens. Colitis ulcerosa ist
auf Rektum und Colon begrenzt und betrifft nur Mucosa und Submucosa;
Perforationen sind selten. Morbus Crohn kann dagegen jeden Abschnitt
des Gastrointestinaltrakts befallen. Häufig betrifft er mehrere Abschnitte
zugleich, z. B. terminales Ileum und Rektum, mit einem Entzündungsprozess,
der in der Regel die gesamte Darmwand einbezieht.
IBD ist allem Anschein
nach eine überschießende Immunreaktion gegen Teile der Darmflora. In
diesem Sinn ist es keine Autoimmunerkrankung, da kein Autoantigen vorliegt.
Andererseits könnten wir unsere Darmmikrobiota mit einiger Berechtigung als
Teil unseres "weiteren Selbst" betrachten, da wir dafür unter
normalen Umständen Immuntoleranz entwickeln. Bei Patienten mit IBD hat
dieser Tolerisierungsprozess nicht ausreichend funktioniert.
Dieser mangelnden Tolerisierung wird offensichtlich durch bestimmte Allele polymorpher Genloci Vorschub geleistet. Bei vielen IBD-Patienten ist die Barrierefunktion des Darmepithels beeinträchtigt:
- Während der innere Zusammenhang noch nicht geklärt ist, findet man bei Patienten mit bestimmten NOD2-Allelen, dass die tight junctions zwischen Enterozyten durchlässiger sind als gewöhnlich. Erinnern wir uns: NOD2 ist ein intrazellulärer pattern recognition receptor für PAMPs (pathogen-associated molecular patterns) aus der Bakterienzellwand. Erinnern wir uns auch, dass H. Pylori diesen Mechanismus aktiv missbraucht, um die Magenwand aufzulockern.
- Patienten mit spezifischen Allelen anderer Gene produzieren geringere Mengen an Defensinen, antibakteriell wirkenden Peptiden, die z. B. von Paneth-Zellen am Boden der Krypten freigesetzt werden.
- Wieder andere Patienten haben einen veränderten IL‑23-Rezeptor. IL‑23 ist ein mit IL‑12 verwandtes proinflammatorisches Zytokin, das TH17- und NK-Zellen in ihrer Proliferation und Funktion anregt.
-
Die Barrierefunktion des Darms kann auch durch entzündliche Schädigung von Zellmembranen verschlechtert werden. Langkettige mehrfach ungesättigte Fettsäuren sind, wie wir gesehen haben, besonders anfällig für oxidative Veränderungen durch reaktive Sauerstoffverbindungen (ROS– reactive oxygen species). Geschützt werden diese Membranfettsäuren durch das Selenoenzym GPX4 (Glutathionperoxidase 4). Menschen mit genetisch bedingt schwächerer GPX4-Funktion weisen nicht nur eine veränderte Darmmikrobiota sondern auch eine verstärkte Neigung zu Darmentzündung über diesen Mechanismus auf, wenn auch die genauen Zusammenhänge erst erforscht werden müssen. Für diese Patienten ist es also ratsam, den Konsum von PUFA zu reduzieren.
Zusammengefasst wurden mehr als 250
mit der Erkrankung assoziierte polymorphe Loci identifiziert, von denen einige
die Gemeinsamkeit haben, dass sie das Eindringen vermehrten bakteriellen
Materials in die Darmwand plausibel erscheinen lassen, andere, dass sie
überschießende Reaktionen des Abwehrsystems begünstigen.
Zellen des adaptiven Immunsystems
reagieren auf dieses bakterielle Material. Dendritische Zellen aktivieren
naive T-Zellen zu TH1 und TH17-Zellen. TH17-Zellen verstärken frühe
nicht-adaptive Abwehrmechanismen, z. B. die Rekrutierung von neutrophilen
Granulozyten. Granulozyteninfiltrate mit Kryptenabszessen sind typisch
für IBD. Makrophagen werden sowohl durch PAMPs direkt, als
auch durch TH1-Zellen via IFNγ aktiviert. Durch Makrophagen und
TH1-Zellen sezerniertes TNFα trägt durch Induktion von Proteasen
zur Gewebsdestruktion bei, und die Proteasen schädigen ihrerseits die
tight junctions des Epithels. In einem
Circulus vitiosus verstärken also unspezifische
und adaptive Abwehrmechanismen die Durchlässigkeit des Darmepithels.
Pharmakologische Querverstrebung: Anti-TNFα-Therapie hat einen positiven Effekt bei
fistelbildendem Morbus Crohn. Anti-IL‑23-Therapie ist gegen beide Erkrankungen zugelassen: Einerseits Ustekinumab, ein
Antikörper, der die gemeinsame Betakette ("p40") von IL‑12 und
IL‑23 bindet und damit die Aktivierung ihrer jeweiligen Rezeptoren
verhindert. Weiters Risankizumab,
der die Alphakette von IL‑23 ("p19") bindet.
Umweltfaktoren haben ebenfalls
einen Einfluss. In einer Parallele zur Allergie weisen Populationen,
die bestimmten Infektionen weniger ausgesetzt sind, eine höhere Morbidität
durch IBD auf. Die Hygienehypothese postuliert eine Notwendigkeit für
Infektionen zur Etablierung eines normalen Toleranzniveaus. Es gibt
Tiermodelle, bei denen Wurminfektionen die Entstehung von IBD verhindern
oder das Krankheitsbild abschwächen. Passend zu dieser Hypothese steigt
die Häufigkeit von IBD weltweit an, mit der höchsten Prävalenz in Europa,
Nordamerika und Australien.
Überschießende Immunreaktionen
zeigen sich auch in extra-intestinalen Manifestationen, wie z. B. Erythema nodosum, Polyarthritis migrans, Sacroileitis, Morbus Bechterew, Uveitis oder
Cholangitis.
Die Krankheitserscheinungen
sind abhängig von Lokalisation und Intensität und sehr variabel. Morbus
Crohn beginnt häufig mit mäßigem Durchfall, Bauchschmerzen und Fieber
und kann so eine Appendizitis vortäuschen. Colitis ulcerosa, die Rektum
und anschließende Teile des Colons befällt, beginnt oft mit Krämpfen
im Unterbauch und blutig-schleimigen Durchfällen. Beide Erkrankungen
tendieren dazu, in jungen Jahren zu beginnen und zeigen einen chronischen,
durch wiederkehrende Episoden gekennzeichneten Krankheitsverlauf.
19. PATHOPHYSIOLOGIE DER LEBER
Obwohl sich der menschliche Organismus
von der Außenwelt durch deutliche Grenzen absetzt, ist natürlich ein
Stoffaustausch mit der Umwelt unabdingbar. Dieser birgt erhebliche Risiken:
einerseits können sorgsam regulierte Gleichgewichtszustände
("Homöostasen") durcheinander geraten, andererseits können toxische
Substanzen in den Körper gelangen. Der zur Vermeidung solcher Störungen
zuständige "Manager" ist die Leber.
Um ausreichend Nahrungsstoffe aufnehmen
zu können, wird eine große Austauschfläche –die Darmoberfläche—benötigt. Eine
sofortige Bearbeitung und Kontrolle der aufgenommenen Stoffe beim Durchtritt
durch das Darmepithel wäre wohl "technisch" zu aufwendig. Diese
erfolgt also später, nachdem das von dieser riesigen Austauschfläche stammende
Blut wieder gesammelt wurde und durch die Enge "Pforte" der Leber
geschleust wurde. Es werden also zwei Kapillarsysteme in einem
Niederdrucksystem hintereinander geschaltet. In der Leber müssen erneut große
Mengen von Molekülen zwischen Blut und Hepatozyten ausgetauscht werden. Das
wird erleichtert durch eine Strömungsverlangsamung durch die breiten
Lebersinusoide und durch die Fenestrierung des Leberendothels, die dem
Blutplasma direkten Zugang in den dahinterliegenden Disse´schen Raum erlaubt,
in den die Transporter-Protein-besetzten Mikrovilli der Hepatozyten ragen. Im
Disse´schen Raum befinden sich außerdem spezialisierte Perizyten, die
Sternzellen (hepatic stellate cells)
oder Ito-Zellen, welche Vitamin A speichern und Bestandteile der
extrazellulären Matrix synthetisieren können. Diese ist bei einer gesunden
Leber sehr zart ausgebildet, um die Diffusionswege zu minimieren. In direktem
Kontakt mit dem Blut, also innerhalb der Sinusoide, sitzen zwei Zellarten der
unspezifischen Abwehr: eine große Zahl von Makrophagen, die Kupffer-Zellen,
sowie NK-(natural killer) Zellen, die
sogenannten Pit-Zellen.
Aus der Rolle der Leber in der
Auseinandersetzung mit der Außenwelt ergibt sich auch die Notwendigkeit,
Moleküle zurück in den Darm zu schicken. Dies wird durch das Gallengangssystem
inklusive Gallenblase erreicht. Die kleinsten Verästelungen der Gallengänge
beginnen als Spalträume zwischen benachbarten Leberzellen und werden nur durch
deren Zellmembran begrenzt. Diese Canaliculi bilden ein dreidimensionales
polygonales Netzwerk mit zahlreichen Anastomosen. Erst die größeren Ductuli haben
ein eigenes Epithel.
Welche Bedeutung das Funktionieren der
Leber für den Organismus hat, zeigt sich bei einem plötzlichen Ausfall der
Leberfunktion z. B. in Folge einer Knollenblätterpilz-(Amanita phalloides)-Vergiftung. Das
Hauptgift, Amanitin, ein zyklisches Peptid aus 8 Aminosäuren, dessen Struktur
so stabil ist, dass sie durch Kochen nicht verändert wird, bindet an die 140
kDa-Untereinheit der RNA-Polymerase II. Die
so blockierte Genexpression führt zur Nekrose der Hepatozyten und kann innerhalb
von 48-72 Stunden zum Tod des Patienten führen.
Im Folgenden werden die Funktionen der
Leber kurz besprochen, woraus sich die klinischen Folgen von Funktionsstörungen
meist ableiten lassen.
Funktion: Energiestoffwechselhomöostase
Störung: Schwäche, Fettstoffwechselstörungen, nicht-alkoholische Fettlebererkrankung, Insulinresistenz, metabolisches Syndrom
Zeiten der Nahrungsaufnahme (Resorptionsphase) wechseln ab mit Zeiten ohne Energieträgerangebot aus dem Darm (Postresorptionsphase), eventuell sogar mit Hungerphasen. Die Leber hat wichtige Pufferfunktionen zur Erhaltung eines konstanten Energieträgerangebots im Blut.
In der Resorptionsphase wird viel Glucose über den Darm aufgenommen, die Gluconeogenese wird daher gehemmt: Insulin bewirkt in Hepatozyten eine Phosphorylierung und den Abbau des Transkriptionsfaktors Foxo1. Foxo1 treibt sonst die Transkription von Enzymen der Gluconeogenese (PEPCK, Glucose-6-Phosphatase). Ein Teil der im Darm resorbierten Glucose wird mit Hilfe des insulinunabhängigen Transporters GLUT2 (KM 15-20 mM) in die Hepatozyten transportiert. Glucose kann als solche nicht gespeichert werden, doch kann die Leber –wie auch die Skelettmuskeln—Glucose zu Glykogen polymerisieren. Glykogen kann bis zu 10% des Lebergewichts ausmachen (100-120g). Die Menge an Energie, die in dieser Form gespeichert werden kann, ist jedoch sehr begrenzt: die vielen Hydroxylgruppen der Glucoseeinheiten wirken stark hydrophil: 1g Glykogen bindet 2.7 g Wasser. Diese Form der Energiespeicherung nimmt zu viel Volumen und Gewicht in Anspruch, um effizient zu sein. Überschüssige Glucose wird deshalb in der Leber über Acetyl-CoA zu Fettsäuren umgebaut; diese werden mit Glycerol zu Triglyceriden vereinigt und in Form von VLDL (very low density lipoprotein)-Partikeln großteils an das Blut abgegeben. Werden allerdings Kohlenhydrate mit der Nahrung über die Zeit zu häufig oder in zu großen Mengen zugeführt, steigt die Konzentration von freien Fettsäuren und Fetten in den Hepatozyten so weit an, dass es zu Funktionseinschränkungen, beginnend mit Insulinresistenz kommt (MASLD, metabolic dysfunction–associated steatotic liver disease, früher NAFLD, non alcoholic fatty liver disease). Dieser proinflammatorische Effekt kann so stark werden, dass es zu Zelluntergang (Steatohepatitis) und Vernarbung/Fibrose kommt. Die dritte Aufgabe der Leber in der Resorptionsphase ist die Aufnahme von Chylomikronenremnants, die Bearbeitung der in ihnen enthaltenen komplexen Lipide, darunter z. B. fettlösliche Vitamine oder Fremdstoffe, sowie die Wiederabgabe behandelter Lipide in der Form von VLDL. Aufgenommene Fette sind ja das einzige Segment der Nahrung, das die Leber zunächst umgeht. Das Darmepithel formt diese zu Chylomikronen um und setzt diese in den extrazellulären Raum frei, von wo sie mit der Gewebslymphe in die Lymphbahnen gespült werden. Von dort gelangen sie im Venenwinkel ins Blut. Die Triglyzeride werden mit Hilfe der Lipoproteinlipase auf der Endothelmembran von Muskel- und Fettgewebe aus den Chylomikronen geholt; erst die Remnants werden von der Leber aufgenommen.
In der Postresorptionsphase werden unter Kontrolle von erniedrigtem Insulin und Leptin sowie erhöhtem Glucagon und Sympathikotonus die Energiespeicher angezapft, um einen kontinuierlichen Energieträgerspiegel im Blut sicherzustellen. Fettsäuren aus Depotfett sind in großen Mengen verfügbar, doch nicht alle Zellen sind in der Lage, diese zu verstoffwechseln. Für manche Gewebe (ZNS, Erythrozyten, Nierenmark) ist Glucose unentbehrlich. Das Muskelglykogen steht dafür nicht zur Verfügung, es kann nur im Muskel selbst, z. B. zu Laktat, verbraucht werden. Im Blut wird ein langsam sinkender Glucosespiegel zunächst hauptsächlich durch Glykogenolyse, Abbau des Leber-Glykogens aufrechterhalten.
Parallel steigt die Gluconeogenese, die Neusynthese von Glucose aus Laktat, Glycerol und Aminosäuren. Dies wird über zwei Wege reguliert:
- Der sinkende Insulinspiegel enthemmt in den Hepatozyten die Foxo1-abhängige Transkription von Enzymen der Gluconeogenese.
-
Sinkender Blutzucker- und Insulinspiegel hemmen in Fettzellen die Ausschüttung von Leptin. Das ZNS reagiert auf diesen Leptinrückgang mit einer Aktivierung der "Stress-Achse" CRH-ACTH-Cortisol (typischer Höchstwert von Cortisol in den frühen Morgenstunden nach der Nahrungskarenz über Nacht; zur selben Zeit erfolgt die pulsatile Ausschüttung von Wachstumshormon). Cortisol und Wachstumshormon induzieren die lipolytischen Enzyme adipöse Triglyceridlipase (ATGL) und hormonsensitive Lipase (HSL) sowie β-Adrenozeptoren, sodass die Ansprechbarkeit für Adrenalin/Noradrenalin erhöht wird. Cortisol, Wachstumshormon, Glucagon, ACTH und Sympathikusaktivierung werfen nun in Zusammenarbeit mit dem nun niedrigen Insulinspiegel die Lipolyse im Fettgewebe an. Aus den Fettzellen mobilisierte freie Fettsäuren (free fatty acids, FFA, oder non-esterified fatty acids, NEFA) und Glycerol steigen im Blut an und erreichen die Leber. Hepatozyten bauen diese Fettsäuren zu Acetyl-CoA ab; Acetyl-CoA aktiviert allosterisch das Enzym Pyruvatcarboxylase, das die erste Reaktion der Gluconeogenese (zu Oxalazetat) katalysiert. Die Lipolyse im Fettgewebe bewirkt so über einen erhöhten Umsatz der Leber-Pyruvatcarboxylase eine verstärkte Gluconeogenese.
Das dazu benötigte Ausgangsmaterial
stellen neben begrenztem Glycerol und Laktat vor allem Aminosäuren dar, die ebenfalls
Cortisol, der Dirigent des Stoffwechselorchesters, gleichzeitig aus Muskeln und
Knochen mobilisiert. Aus Muskeln und Knochen?? Aber die brauchen wir doch?!
Pfff..., wir haben solche Unmengen von Protein in Muskeln und Knochen, dass wir
kurzzeitig problemlos diesen winzigen Bruchteil für die Gluconeogenese
"ausleihen" können, in der Erwartung, dieses überzogene Konto bald
wieder ausgleichen zu können. Durch gefinkelten
Aminosäuremetabolismus in der Muskelzelle liefern die Muskeln als Standardenergieträger
hauptsächlich Alanin an die Leber. Alanin-Aminotransferase (ALT oder GPT,
Glutamat-Pyruvat-Transaminase) wandelt dort Alanin + α‑Ketoglutarat
zu Pyruvat + Glutamat um. Pyruvat wird in die Gluconeogenese eingeschleust, der
Glutamat-Stickstoff muss entsorgt werden.
Behalten wir im Gedächtnis, dass für beide Wege ein sehr niedriger Insulinspiegel Voraussetzung ist. Steigt der Insulinspiegel geringfügig, senkt das die Gluconeogeneserate. Machen wir uns ebenfalls noch einmal bewusst, dass Insulin die Lipolyse blockiert. Wenn wir also abends vor der Glotze noch eine Süßigkeit schnabulieren oder nachts hungrig den Kühlschrank ansteuern, verhindern wir die für diese Zeit vorgesehene Lipolyse; wir aktivieren wieder die Resorptionsphase und synthetisieren Fett, statt es abzubauen.
Pharmakologische Querverstrebung: Pharmakologische Dosen von Glucocorticoidenbewirken einerseits eine starke Erhöhung des
Blutzuckers – Ankurbelung der Gluconeogenese – und wirken andererseits peripher
katabol – als Material für die Gluconeogenese benötigen wir ja Aminosäuren, die
wir aus Knochen und Muskel entnehmen –, sodass bei längerer Therapiedauer
Osteoporose und Verlust an Muskelmasse auftritt.
Hungerphase. Dauert die Phase ohne Nahrungszufuhr oder
mit zu geringer Nahrungszufuhr länger als einen Tag an, ist das Leberglykogen
verbraucht. Zusätzlich geht die Anlieferung von Alanin aus dem Muskel und damit
die Gluconeogeneserate zurück: Der Blutzucker sinkt von etwa 90 auf etwa 70
mg/dl. Wie diese Regulation erfolgt, ist noch nicht ausreichend klar, sie wird
jedenfalls vom ZNS organisiert. Es halbiert sich der Leptinspiegel, TSH geht
noch stärker zurück, mit dem dadurch reduzierten Schilddrüsenhormon reduziert
sich auch der Gesamtenergieverbrauch: so wird z. B. die mitochondriale
Energieerzeugung der Leber auf Sparflamme gesetzt. Man beginnt zu frieren, die
Durchblutung der Haut nimmt ab, besonders Hände und Füße fühlen sich eiskalt
an. Das reduziert Wärmeverluste nach außen und spart damit ATP-Heizkosten ein. Auch
der Energieverbrauch des Herzens wird in Ruhe reduziert: das Herz schlägt etwas
langsamer, der Blutdruck sinkt etwas ab. Während sich der Körper in der
Postresorptionsphase kurzfristig ebenso hemmungs- wie gefahrlos an den Muskeln bedienen
konnte, muss er nun seine Strategie umstellen: Die Muskeln kann er nicht
längerfristig kannibalisieren, da sie weiter zur Nahrungssuche etc. benötigt
werden.
Gleichzeitig stellt die Leber aus den immer
stärker anflutenden Fettsäuren zusätzlich so genannte Ketonkörper, β-Hydroxybutyrat
und Azetoazetat, her. Das geschieht dadurch, dass die Leber mehr angelieferte
Fettsäuren durch β-Oxidation abzubauen beginnt, als sie via
Acetyl-CoA über den Zitratzyklus vollständig verstoffwechseln kann.
Überschüssiges Acetyl-CoA führt bei Glucoseüberschuss zu Fettsäuresynthese, wie
wir gesehen haben, doch bei Glucosemangel zur Produktion von Ketonkörpern. Um
das Acetyl-CoA in den Zitratzyklus einzubringen und zu "verbrennen",
ist nämlich Oxalazetat notwendig ("fats
burn in the flame of carbohydrates"), das nun auch für Gluconeogenese
verbraucht und damit limitierend wird., Da, bildlich gesprochen, nicht mehr
alles im Kraftwerk abgearbeitet werden kann, beginnt sich der
Acetyl-CoA-Stausee zu füllen, bis der Spiegel das Niveau des Überlaufs
erreicht: Jeweils zwei der gestauten Acetyl-CoA werden zusammengehängt (4
Kohlenstoffe; Reaktion über HMG-CoA), von CoA abgehängt und hauptsächlich in Form
von β‑Hydroxybutyrat, zu einem kleineren Teil als Azetoazetat ins
Blut abgegeben. Die beiden sind
mittelstarke Säuren, geben also Protonen ab, es entsteht eine ketoazidotische
Stoffwechsellage. Dabei übernimmt die Nierenrinde bis zu einem Drittel der nun reduzierten
Gluconeogenese: Erinnern wir uns, dass der proximale Tubulus Säureausscheidung in Form von NH4+ mit Gluconeogenese verbindet. Herz und Nierenrinde verbrennen ohnehin
gerne Ketonkörper; Großenergieverbraucher ZNS, das mit Fettsäuren wenig
anfangen kann, kann sich nach einiger Zeit daran gewöhnen und verstoffwechselt dann
bevorzugt Ketonkörper (bis zu 75% des Energiebedarfs) neben Glucose. (Anmerkung:
Traditionelle Ansicht ist, dass das ZNS Fettsäuren nicht verstoffwechseln kann.
Neuere Daten widersprechen dem und weisen Fettsäureverbrennung in Mitochondrien
in der Nachbarschaft von aktiven Synapsen nach. Quantitativ ist dieser Anteil
an der Gesamtenergiegewinnung des ZNS wahrscheinlich aber nicht sehr groß.) Angesichts
des Energiehungers unseres Gehirns kann so in längeren Hungerphasen viel
Glucose eingespart werden. Ohne diese Umstellung würden in anhaltenden Hungerperioden
die Muskeln viel zu rasch abgebaut werden: der Proteinverbrauch fällt von ca.
75g pro Tag auf 20g. Die Ketose ist also im Grunde ein Trick, um das ZNS von
den Fettspeichern zu ernähren: das Fettgewebe setzt Fettsäuren frei, die von
der Leber zu Ketonkörpern umgebaut werden, die ihrerseits zur Hauptnahrung des ZNS
werden. Gleichzeitig entsteht durch die erhöhten Fettsäurekonzentrationen Resistenz
gegen das ohnehin schon sehr niedrige Insulin in den typischen von Insulin
gesteuerten Geweben Muskel, Fettgewebe, Leber, sodass von der ohnehin nur mehr spärlich
produzierten Glucose ein größerer Anteil für ZNS, Erys und Nierenmark übrig
bleibt.
Möglicherweise ist also die durch erhöhte Konzentration freier Fettsäuren ausgelöste Insulinresistenz, die wir lange als ausschließlich pathologisches Phänomen betrachtet haben, ein physiologischer Bestandteil eines Notfallprogramms, das unseren Vorfahren ermöglicht hat, lange Hungerphasen mit sehr niedrigen Blutzuckerspiegeln zu überleben. Anhaltender Nahrungsüberfluss ist in der menschlichen Evolution bisher nicht aufgetreten. Heute wird uns dieser Mechanismus zum Verhängnis. Bei starkem Übergewicht mit metabolischem Syndrom haben wir zwar die gegenteilige Ernährungslage – üppig statt karg –, jedoch ebenso eine erhöhte Konzentration freier Fettsäuren, die nun zu Insulinresistenz bei sehr hohen Blutzuckerspiegeln führt.
Der Zustand der Ketose wird also nur langsam, nach mehreren Tagen, erreicht. Im
Gegensatz dazu wird er sofort unterbrochen, wenn wieder exogene Kohlenhydrate
verfügbar werden: die Kohlenhydrate induzieren einen Insulinpeak, und Insulin
bremst die Lipolyse rasch und anhaltend. Wenn immer er kann, schont unser
Organismus seine Energiereserven: "Wenn es heute Kohlenhydrate gibt,
stehen die Chancen gut, dass es morgen auch welche geben wird!". Fallen die
Kohlenhydrate wieder aus, dauert es erneut mehrere Tage, bis der Zustand der
vorwiegenden Fettverbrennung/Ketose wieder erreicht wird.
Der metabolische Zustand der Ketose ist
nicht gleichbedeutend mit einer Reduktion der Fettreserven. Das ist nur bei
einem ernährungsbedingten Kaloriendefizit der Fall. Ketose bedeutet lediglich
den metabolischen Zustand der vorwiegenden Fettverbrennung. Bei reichlicher
Zufuhr von fett- und proteinreichen Nahrungsmitteln, also einem
Kalorienüberschuss ohne Kohlenhydrate, ist es möglich, in einem Zustand der Ketose
die bestehenden Fettpölster sogar noch auszuweiten.
Ernährung: besser Kohlenhydrate oder besser Fett?
Ketogene Diät. Der
Proteinanteil in der Ernährung liegt typischerweise bei 16-18%, bei den
allermeisten fällt er in den Bereich 10-35 %. Ein Proteinanteil über 35%
ist kaum zu essen und aus mehreren Gründen nicht ratsam. So belasten wir unsere
Nieren umso stärker, je mehr Protein wir essen. Den Rest können wir zwischen
Kohlenhydraten und Fett verteilen. Die gebräuchliche Empfehlung lautet
45-65 % Kohlenhydrate, 20-35 % Fett. Eine ketogene low carb Ernährungsweise reduziert den
Kohlenhydratanteil extrem zugunsten des Fettanteils. Damit wird die Hungerphase
in manchen Aspekten imitiert. Durch die stark reduzierte Insulinausschüttung
kann diese Ernährungsart in Verbindung mit Kalorienreduktion helfen, das
Gewicht langsam zu reduzieren. Nach einer Übergangsphase, in der viel Wasser
verloren wird (da die Glykogenvorräte abgebaut werden; sieht auf der Waage
super aus: "Wow, endlich eine funktionierende Diät, ich habe schon 2 kg
abgenommen!"), kann sich der Körper erstaunlich gut auf diese
Ernährungslage einstellen. Manche Populationen, wie die Inuit, haben sich
mangels Kohlenhydraten systematisch so ernährt: Fische und Robben enthalten
wenig Kohlenhydrate, aber reichlich Fette und Proteine. Für eine kurz- bis
mittelfristige Zeit kann eine solche Diät helfen, das Gewicht zu reduzieren.
Wie sieht es langfristig aus?
Langfristige
ketogene Ernährung? Seit langer
Zeit schon wissen wir, dass sich ketogene Ernährung positiv auf bestimmte
Epilepsieformen auswirkt. Leider
ist die Datenlage zur Frage, wie gesund oder ungesund unterschiedliche Verteilungen
der Energieträger Kohlenhydrate, Fett und Protein sind, sehr unzureichend. Im
Idealfall wünschen wir uns randomisierte kontrollierte Studien über dreißig und
mehr Jahre mit zigtausend Teilnehmer:innen, in denen die Menschen sich ständig,
genau dokumentiert und kontrolliert an eine vorgegebene Verteilung der
Makronährstoffe halten, natürlich unter Einhaltung aller Erfordernisse für Mikronährstoffe
(Mineralstoffe, Vitamine, Spurenelemente). Das alles gekoppelt mit minutiöser
Dokumentation der Entwicklung ihrer Gesundheit. Abgesehen davon, dass das nicht
möglich ist: Würden Sie bei so einer Studie mitmachen? Dreißig Jahre keine Süßigkeiten,
weil Sie in die Ketogruppe randomisiert wurden? Es gibt aus diesem Grund sehr
wenige Studien, die einigermaßen belastbare Daten zu unserer Frage bieten. In
diesen Studien wurden die Teilnehmer:innen am Beginn, eventuell noch einmal
später, zu ihren Ernährungsgewohnheiten per Fragebogen befragt. Dabei entstehen
bereits reichlich Unschärfen. Diese Angaben wurden dann per
Standardumrechnungen – wieviel kcal aus Kohlenhydraten, Fett und Protein enthält
eine Portion Lauch? ein Marmeladebrot? eine Pizza? – in eine
Makronährstoffverteilung umgerechnet. Dann nahm man an, dass sich diese Ernährungsgewohnheiten
über die Studienjahre nicht wesentlich änderten. Sie sehen schon: diese Daten
enthalten zahlreiche Ebenen der Unsicherheit. Die aussagekräftigsten Daten
kommen meines Erachtens aus drei prospektiven Studien: PREDIMED (Spanien), PURE
(18 Staaten mit dem Hauptgewicht in Asien und Südamerika) und ARIC (4 Einzugsgebiete
in den USA):
- PREDIMED ist die einzige randomisierte interventionelle Studie von den dreien. Drei Gruppen von Probanden mit kardiovaskulärem Risiko wurden verglichen: basierend auf mediterraner Kost wurde die erste ermutigt, vermehrt kaltgepresstes Olivenöl zusätzlich zu konsumieren; die zweite, täglich Nüsse (Mandeln, Hasel- und Walnüsse, reich an mehrfach ungesättigten Fettsäuren) zu ergänzen (Öl bzw. Nüsse wurden zur Verfügung gestellt). Die dritte Gruppe sollte sich bemühen, Fett etwas zu reduzieren – was automatisch eine Vermehrung der Kohlenhydrate bedeutet – und erhielt statt Öl/Nüssen kleine non-food Geschenke. Der zusammengesetzte Endpunkt beinhaltete Myokardinfarkt, Schlaganfall, Tod aus Herz-Kreislaufgründen. Ergebnis: die fettreduzierte Gruppe hatte ein klar erhöhtes Risiko gegenüber den beiden Gruppen, die vermehrt pflanzliche Fette konsumiert hatten.
-
Die Hauptaussage von PURE zu dieser Fragestellung:ein höherer Fettanteil in der Ernährung ist nicht, wie lange angenommen, mit erhöhter, sondern mit gesenkter Mortalität assoziiert. Allerdings gilt diese Aussage nur für den Kohlenhydrat-Spektrumsanteil "mittel" (niedrigstes Fünftel der Teilnehmenden: 46 % Kohlenhydrate) bis hoch (höchstes Fünftel: 77 %), geringere Kohlenhydratanteile waren nicht vertreten. Für diesen mittleren bis hohen Spektrumsanteil ergab sich: je mehr Kohlenhydrate, desto höher die Mortalität.
-
Nur die ARIC-Studie enthielt einen Anteil an Probanden, die weniger als 46 % Kohlenhydrate zuführten. Hier ergab sich eine U-förmige Mortalitätskurve mit einem Minimum bei einem Kohlenhydratanteil von 50-55%; darüber und darunter stieg die Mortalität. Die Probanden mit der niedrigsten Kohlenhydratzufuhr ernährten sich also in erster Linie von Fett. Das bedeutete in der Regel eine hohe Zufuhr von gesättigtem, tierischem Fett und eine deutliche Zunahme der Mortalität. Ein kleiner Teil ernährte sich vorwiegend von pflanzlichen – ungesättigten – Fetten und Proteinen. Dieser Teil zeigte eine erniedrigte Mortalität, doch ist der Anteil zu gering, als dass wir sichere Schlüsse ziehen könnten.
Was lernen wir daraus? Alle drei Studien sprechen gegen eine fettreduzierte Ernährung, wie sie jahrzehntelang als ideal gepriesen wurde. Über kohlenhydratreduzierte Ernährung geben PREDIMED und PURE keine Auskunft; ARIC findet ein Mortalitätsminimum bei 50-55 % Kohlenhydratanteil, darunter stieg die Mortalität wieder. Zu ketogener Ernährung haben wir bisher keine belastbaren Daten. So niedrige Kohlenhydratzufuhr, wie sie eine ketogene Ernährung erfordert, kommt auch in der ARIC-Studie nicht vor. Aus den ARIC-Daten kann man aber extrapolieren, dass ketogene Ernährung auf die Dauer wohl umso weniger gesund ist, wenn die Energiezufuhr hauptsächlich auf tierischen Fetten und Proteinen basiert (zumindest, wenn diese nicht gerade ausschließlich aus Omega-3-reichen Fettfischen wie vormals bei den Inuit stammt). Eine Unsicherheit besteht bezüglich ketogener Ernährung auf pflanzlicher Basis. Wir können nicht ausschließen, dass eine solche gesund sein KÖNNTE, doch fehlen uns bisher die Daten. Sie ist aber praktisch nicht leicht zu bewerkstelligen (pure peanut butter, anybody? Tofuflößchen in Olivenöl?).
Ist das nicht ein Widerspruch zur Erwartung, dass sich auch beim Menschen eine langjährige Nahrungseinschränkung lebensverlängernd auswirkt? Ist es nicht auch eine ketogene Ernährung, wenn man sich jahrelang hypokalorisch ernährt? Nein: hypokalorisch ernähren ist nicht fasten. Ernährt man sich hypokalorisch, aber bezüglich Makronährstoffen ausgewogen, entsteht keine Ketose, da regelmäßig Kohlenhydrate aufgenommen werden und der Körper nicht auf vorwiegende Fettverbrennung umstellen muss.
GLUT1-Defizienzsyndrom. Ein extrem seltener genetischer Defekt von GLUT1
manifestiert sich vor allem im ZNS. Die Neuronen bekommen zu wenig Energie, was
bei den betroffenen Kindern zu Krämpfen führt und im Lauf der Zeit zu
Entwicklungsstörungen. Welchen alternativen Brennstoff kann man den Neuronen
zuführen? Ketonkörper, die zu allen Zeiten in einem bestimmten Konzentrationsbereich
angeboten werden müssen. Das bedeutet eine außerordentlich konsequente, rein
ketogene Ernährung für das ganze Leben. Die Betroffenen dürfen nur minimale
Mengen an resorbierbaren Kohlenhydraten zu sich nehmen, da die Leber die
Ketonkörperproduktion einstellt, sobald der Blutzuckerspiegel etwas ansteigt.
[Für besonders Interessierte:
Auf welche Weise bremst ein Blutzucker-/Insulinanstieg die Ketonkörperproduktion? Das Kommen und Gehen der Ketose ist asymmetrisch: nach fehlender Kohlenhydratzufuhr wird Ketose nur nach Tagen erreicht; werden dann Kohlenhydrate wieder zugeführt, beendet das die Ketose sofort. Das geschieht auf mehreren Wegen. Die raschesten Wege benötigen keine Änderung der Genexpression. Betrachten wir zwei davon, welche die Ketogenese innerhalb von 1-2 Stunden stilllegen:
1. Das "Gaspedal" der Lipolyse ist die cAMP-gesteuerte PKA-Aktivität.
- Wer tritt auf das Gaspedal? Eine Reihe von Stresshormonen: cAMP wird in Fettzellen durch Gs-Protein-gekoppelte β3-Rezeptoren für Adrenalin/Noradrenalin – also Sympathikus-aktivierung –, Glucagon und ACTH gesteigert.
- Was bewirkt das Gaspedal? Lipidtröpfchen in weißen Fettzellen sind mit dem Protein Perilipin‑1 bedeckt, das einerseits den Zugriff der Lipasen auf die Triglyceride behindert, andererseits das Protein CGI‑58 bindet und damit inaktiviert. PKA-Phosphorylierungen führen zu einer Konformationsänderung von Perlipin, sodass es die Lipasen nun durchlässt. Außerdem lösen die Phosphorylierungen CGI-58 von Perilipin-1 ab, sodass es als Cofaktor für die adipöse Triglyceridlipase (ATGL) fungieren kann. ATGL schneidet die erste Fettsäure aus den Triglyceriden heraus. PKA-Phosphorylierungen aktivieren außerdem die hormonsensitive Lipase, welche die zweite Fettsäure herauslöst. Die Fettsäuren werden via Blut zur Leber transportiert. Der Fettsäure-"Stausee" des Körpers wird langsam aufgefüllt. Wird der Andrang von Fettsäuren allmählich zu groß, um vollständig verstoffwechselt zu werden, beginnt in der Leber die Ketonproduktion.
Was bewirkt kurzfristige Kohlenhydratzufuhr? Innerhalb von Minuten einen Insulinpeak. Insulin aktiviert in den Zielzellen AKT (Proteinkinase B). AKT aktiviert Phosphodiesterase 3B, die cAMP abbaut. Das Gaspedal PKA wird also stillgelegt, die Lipolyse und damit die Versorgung der Leber mit Fettsäuren kommt innerhalb von kurzer Zeit zum Erliegen.
2. Ein weiterer Weg ist eine direkte Hemmung der ß-Oxidation, sobald der Blutzucker geringfügig ansteigt: Insulin aktiviert in Leberzellen die Proteinphosphatase 2A. Diese aktiviert Acetyl-CoA-Carboxylase durch Entfernung eines hemmenden Phosphats. Dieses Enzym setzt also nun mehr Acetyl-CoA zu Malonyl-CoA um. Malonyl-CoA hemmt allosterisch Carnitin-Palmitoyltransferase 1, die für den Transport von Fettsäuren ins Mitochondrium verantwortlich ist. Gelangen weniger Fettsäuren ins Mitochondrium, versiegt das Material für die β-Oxidation und damit auch der Anstau von Acetyl-CoA, der die Ketogenese treibt.]
Diabetes Mellitus Typ 1. Vor Insulin als Medikament eingesetzt werden konnte, war DM1 ein Todesurteil. Die Abwesenheit von Insulin induziert die ketoazidotische Hunger-Stoffwechsellage. Bei hohem Blutzucker starben die Kinder vor vollen Töpfen an "innerem Verhungern". Sie bauten so lange Muskeln und Substanz ab, bis das mit dem Leben nicht mehr vereinbar war.
Unter dem niedrigen Glucosespiegel der Postresorptionsphase (80-100 mg/dl, entspricht 4,5-5,5 mM) werden kritische Gewebe, wie das ZNS, über GLUT1 und GLUT3 kontinuierlich mit Glucose versorgt. Mit ihrer KM von 1 mM arbeiten diese Transporter immer im Sättigungsbereich. In die β-Zellen der Pankreasinseln gelangt dagegen über die wesentlich höhere Schwelle des GLUT2 (KM 15-20 mM) nur wenig Glucose. Wird der Blutzuckerspiegel durch Gluconeogenese leicht erhöht, gelangt proportional etwas mehr Glucose in die β-Zellen, sodass auch etwas mehr Insulin ausgeschüttet wird. In den Inseln hemmt das Insulin die Glucagonausschüttung in den benachbarten α‑Zellen; weniger Glucagon und mehr Insulin gelangen über in die Pfortader ableitende Venolen zur Leber. Diese Insulinwirkung in Kombination mit der verringerten Glucagonwirkung limitiert die Gluconeogenese. Die Glucose-Neuproduktion wird damit dauernd durch eine Insulin-Feedbackschleife gedrosselt.
Ohne diese Drosselung würde die Leber viel mehr Glucose durch Gluconeogenese erzeugen. Genau das geschieht beim Metabolischen Syndrom: hier zeigt die Leber Insulinresistenz und erzeugt mehr Glucose als notwendig. Das erklärt den erhöhten morgendlichen Nüchternzucker der Typ 2-Diabetiker, der sonst unlogisch wäre: warum sollte der Zucker erhöht sein, wenn die Person schon lange nichts mehr gegessen hat? Fazit: Die Leber hat eine enorme Kapazität für Gluconeogenese, die dauernd durch Insulin gedrosselt werden muss.
Pharmakologische Querverstrebung: Metformin hemmt die Gluconeogenese und ist damit das Basismedikament für Diabetes mellitus Typ 2. Im Lauf der Zeit wurden mehrere Mechanismen für diesen Effekt vorgeschlagen. Am überzeugendsten erscheint mir ein Effekt auf ein mitochondriales Redoxenzym, der dazu führt, dass im Zytosol des Hepatozyten mehr NADH und weniger NAD+ bereit steht. Dadurch kann weniger Laktat zu Pyruvat verwandelt und über die Pyruvatcarboxylase in die Gluconeogenese eingeschleust werden. Das erklärt auch eine Tendenz zu Laktatazidose als unerwünschte Nebenwirkung. Wie wir gleich sehen werden, beobachten wir diesen Effekt auch beim Abbau von Alkohol, wodurch die Kombination Metformin + Alkohol besonders problematisch ist.
Funktion: Aminosäuren-Stoffwechsel, Stickstoffausscheidung
(Harnstoffsynthese).
Störung: Hepatische Enzephalopathie, Säure-Basen-Instabilität
Die Gluconeogenese aus Aminosäuren, die
hauptsächlich aus dem Muskel stammen, löst das Problem der Glucoseversorgung in
Hungerzeiten, hat aber mehrere Haken. Zunächst einmal bleibt, wenn man
Aminosäuren zu Kohlehydraten umbaut, der Stickstoff der Aminogruppen übrig. Die
einfachste Form, in der Stickstoff im Körper vorkommt, ist Ammoniak (NH3 bzw. Ammonium-Ion NH4+). Dieses wird allerdings nicht sehr
effizient ausgeschieden und wirkt in höheren Konzentrationen toxisch, was sich
als erstes im ZNS bemerkbar macht.
Die Stickstoffentsorgung beeinflusst auch
den Säure-Basen-Haushalt in unserem Körper, über den wir uns schon im Zuge
unserer Befassung
mit der Niere Gedanken gemacht haben. Dort haben wir vorweggenommen, dass
die für das Säure-Basengleichgewicht kritische Entscheidung, wieviel Stickstoff
in der Form von Harnstoff und wieviel in der Form von NH4+ entsorgt wird,
in der Leber erfolgt. Diese Regulation sehen wir uns nun näher an.
Wieder einmal sitzen wir beim Frühstück
und stellen dabei pathophysiologische Überlegungen an. Proteine sind zwar gute
Energielieferanten, doch ist neben der Stickstoffentsorgungsfrage auch die
Säure-Basen-Situation komplizierter als bei Fetten und Kohlenhydraten. Fette
und Kohlenhydrate (Butterbrot) bauen wir zu CO2 und Wasser ab; CO2 ist eine potentielle Säure, doch die atmen wir ab. 'Wie sieht das eigentlich
bei Proteinen aus?', überlegen wir, während wir unser Frühstücksei genießen.
Die meisten Aminosäuren sind neutral:
sie enthalten zwei gegensätzlich ionisierte Gruppen: eine Carboxyl- und eine
Aminogruppe. Werden sie abgebaut, ensteht (netto, der tatsächliche Abbau ist
natürlich viel komplizierter) also ebensoviel HCO3− wie NH4+.
Eine typische Proteinaufnahme von 100 g/d führt zur Produktion von etwa 1000
mmol HCO3−/Tag und 1000 mmol NH4+/Tag.
Aus der Säure-Basenperspektive könnte NH4+ ein Proton
abgeben, HCO3− eines aufnehmen. Beim
Blut-pH von 7,4 gibt NH4+ mit seinem pKa von
9.2 sein Proton jedoch kaum ab. HCO3− jedoch nimmt
gerne ein Proton auf, um dann als CO2 abgeatmet zu werden, sodass
der Nettoprozess alkalisierend wirken würde.
Beinahe verschlucken wir uns an unserem
Ei: Alarm! NH4+ ist toxisch; HCO3− wirkt alkalisch:
wir müssen etwas tun! Vom Konzept her am einfachsten und logisch wäre es, die
beiden zu einem "Müllmolekül" zu komprimieren. Bingo! Der
Harnstoffzyklus macht genau das. Harnstoff ist, verglichen mit den beiden
schwierigen Charakteren, ein außerordentlich verträgliches Molekül: wenig
reaktiv, untoxisch, unverdächtig aus Säure-Basenperspektive,
Stickstoff-verdichtend. Ein bisschen schwer auszuscheiden, doch da wird unserer
Niere schon etwas einfallen.
Stickstoff-verdichtend? Ja, denn in
Harnstoff kommen zwei NH2-Gruppen auf eine C=O‑Einheit. Hm,
was ist dann mit unserem Säure-Basen-Gleichgewicht? Wenn wir aus NH4+ eine NH2-Gruppe machen, bleibt uns doch ein H+ übrig; na
gut, dem HCO3− fehlt eines, das
gleicht sich vielleicht aus, aber was ist mit dem zweiten NH4+?
Dort bleibt auf jeden Fall ein Proton übrig! (Cave: Wenn wir nicht zugleich
BiochemikerInnen werden wollen, begnügen wir uns damit. Technisch ist alles wie
immer komplizierter; die zweite Aminogruppe stammt nicht direkt aus NH4+ sondern wird von Aspartat aus übertragen. Man kann nun versuchen, die
Stöchiometrien von Reaktion zu Reaktion weiter zu verfolgen. Unterm Strich
jedoch trifft die Aussage zu: Protonen
bleiben übrig.) Resultat: schon wieder beginnen sich unserer Nackenhaare zu
sträuben! Alarm! Haben wir den Teufel mit Beelzebub ausgetrieben? Soeben noch
drohten wir zu alkalisieren, nun drohen wir zu versauern! Was sollen wir tun,
auf Protein verzichten, nur noch Zucker und Fett essen? Schokolade-Diät?
Ahh- Entspannung: da fällt uns ein,
dass unsere Niere Säure ja gut
ausscheiden kann- praktischerweise sogar als NH4+.
Nur müssen wir darauf achten, dass wir die Säureäquivalente in Form von NH4+ nicht direkt auf den Weg von der Leber an die Niere schicken: so viel NH4+ wäre toxisch. Wir benötigen also einen sicheren Säure-/Ammonium-Tankwagen: das
ist Glutamin. Die Niere holt aus diesem Tankwagen wieder das NH4+ hervor und scheidet es aus. Jedes von der Niere ausgeschiedene NH4+ muss nicht mit HCO3− neutralisiert werden und spart
damit HCO3−.
In der Leber haben wir
also zwei Möglichkeiten, mit NH4+ umzugehen:
1.
wir
verwenden es, um das beim Aminosäureabbau entstehende HCO3− zu neutralisieren und stecken es in Harnstoff
2.
wir
stecken es in den Glutamin-Tankwagen, um es in der Niere auszuscheiden
Option 1 verbraucht
HCO3−, Option 2 spart HCO3−.
Wenn es uns nun noch gelingt, das Verhältnis zwischen beiden Wegen intelligent
zu steuern, entkommen wir den beiden Formen der Säure-Basen-Katastrophe, in die
wir durch die Energiegewinnung aus Aminosäuren zu geraten drohen.
Und siehe da, das
Verhältnis zwischen beiden Wegen wird in der Leber durch den pH gesteuert-
intelligenter geht es nicht! Eine geringfügige Senkung des pH reduziert in der
Leber die Harnstoffproduktion, aber steigert die Glutaminsynthese. Damit wird
weniger HCO3− verbraucht und mehr gespart, und die
sich anbahnende Azidose wird ausgeglichen. Umgekehrt hat ein Anstieg des pH die
gegenteiligen Folgen.
[Kein Lernstoff-
Exklusiv für unsere Biochemie-Aficionados:
Die Senkung der
Harnstoffsynthese durch sinkenden pH erfolgt durch das Enzym Glutaminase.
Glutaminase stellt in den Mitochondrien periportaler Zellen das NH4+ zur Verschmelzung mit HCO3− und einem Phosphat aus
ATP zu Carbamoylphosphat zur Verfügung, das anschließend in den Harnstoffzyklus
eingeschleust wird. Diese Leberglutaminase ist in ihrer Aktivität direkt
pH-abhängig: niedrigerer pH → weniger Carbamoylphosphat → weniger
Harnstoffsynthese pro Zeiteinheit.
Einschub Gentherapie:
Das Enzym CPS‑1 (Carbamoylphosphat-Synthetase‑1), das NH4+ mit HCO3− und einem Phosphat aus ATP zu
Carbamoylphosphat komprimiert, was das erste Beispiel für eine erfolgreiche
individuelle Gentherapie. Ein Baby mit massiv erhöhtem NH4+ auf Proteinzufuhr stellte sich als homozygot für eine
Mutation in diesem Gen heraus. Es wurde zunächst durch eine Diät mit stark
reuziertem Proteingehalt am Leben gehalten. Nach wenigen Monaten konnte es
durch ein CRISPR-Cas9-abgeleitetes gene
editing durch Injektion eines Lipid-Nanopartikel-verpackten entsprechenden
Konstrukts mit wesentlich erhöhter Proteintoleranz ausgestattet werden. In
einem beträchtlichen Anteil der Leberzellen wurde das Gen also erfolgreich
repariert.
Die Steigerung der Glutamin-Tankwagenfüllung erfolgt direkt und schlicht durch das Enzym Glutaminsynthase, das im umgekehrten Weg pH-gesteuert wird: niedrigerer pH → mehr Glutamin, das auf die Reise zur Niere geschickt wird. Sinnvollerweise wird dieses Enzym hauptsächlich in Zellen in der Nähe der Zentralvene des Leberläppchens exprimiert: wenn das anfallende NH4+ in der Peripherie des Leberläppchens nicht zu Harnstoff verarbeitet wird, treibt es stromabwärts und muss, vor es die Leber verlässt, im Glutamin-Tankwagen nicht-toxisch transportfähig gemacht werden. ]
Funktion: "Filterung" von partikulärem
Material
Störung: Erhöhte Infektanfälligkeit
Kupffer-Zellen sind eine Form von Makrophagen, die mehr als 80% der
sessilen Makrophagen des retikuloendothelialen Systems darstellen. Sie sitzen
im Inneren der Sinus und phagozytieren sehr effizient partikuläres Material aus
der Pfortader und bauen es ab. Dadurch werden z. B. alternde Erythrozyten
eliminiert, aber auch eingeschwemmte Bakterien inaktiviert. Kupffer-Zellen
exprimieren ein breite Palette an Rezeptoren für PAMPs (pathogen–associated molecular patterns), mit deren Hilfe sie
Bakterien erkennen, phagozytieren und inaktivieren. Sie stellen damit ein
wesentliches Element der unspezifischen Abwehr gegen Infektionen aus dem
Magen-Darm-Trakt dar (siehe Abschnitt über Makrophagen im Immunologie-Skript).
Störung: Vergiftungserscheinungen, abhängig vom spezifischen Molekül.
Über die Darmschleimhaut werden auch viele Stoffe aufgenommen, die eine potentielle Gefahr für den Organismus darstellen. Gerade bei stark lipophilen Molekülen ist es für den Organismus gar nicht so einfach, solche Substanzen wieder loszuwerden. Die Leber hat dazu den Mechanismus der Biotransformation entwickelt, die in zwei Phasen abläuft: in einem ersten Schritt wird, meist durch das Cytochrom-P450-Enzymsystem, eine reaktive Gruppe wie -OH in das Molekül eingebracht, an die in einem zweiten Schritt eine hydrophile Verbindung (z. B. Glucuronsäure, Sulfat, Glutathion etc.) angehängt wird. Das Molekül wird dadurch in der Regel soweit wasserlöslich, dass es über die Galle oder sogar über die Niere ausgeschieden werden kann.
Cytochrom P450-Oxidasen enthalten Häm als eine prosthetische Gruppe, an dessen zentralem Eisen-Atom die eigentliche Redoxreaktion abläuft (der Name stammt von einem Absorptionsmaximum bei 450 nm, wenn CO daran gebunden ist; P-Pigment). Es gibt ca. 50 menschliche Gene für diesen Typ von Enzym, von denen viele in der Leber exprimiert werden. Sie werden nach Familien (Nummern), Subfamilien (Buchstaben) und Genen (Nummern) bezeichnet, z. B. CYP3A4, CYP2D6, CYP2C19, CYP2E1. Eine Reihe von Ursachen hat zur Folge, dass sich einzelne Individuen in ihrer Cytochrom P450-Enzymausstattung unterscheiden:
-
Viele dieser Gene sind polymorph, das bedeutet, verschiedene Individuen haben leicht unterschiedliche Varianten eines solchen Gens und damit des kodierten Enzyms. Die Vielfalt an Genen und Polymorphismen erklärt sich wahrscheinlich aus einem daraus resultierenden Selektionsvorteil in der Menschheitsgeschichte in der Auseinandersetzung mit einer Vielfalt von Alkaloiden aus Nahrungspflanzen.
-
Manche dieser Enzyme werden in Frauen und Männern unterschiedlich exprimiert, z. B. CYP2B13, CYP3A16 und CYP4A12 (Rinn et al., Dev. Cell 6: 791-800, 2004). So wird CYP3A16 nur in der weiblichen, aber nicht in der männlichen Leber exprimiert. Dies kann zu geschlechtsspezifischen Unterschieden in der Metabolisierung von Medikamenten führen.
-
Die meisten dieser Gene werden induziert, wenn das System wiederholt durch ein Substrat beansprucht wird, sodass das Expressionsniveau, abhängig von der individuellen Vorgeschichte, auch bei identer genetischer Situation unterschiedlich sein kann. Mechanismus? Nächster Absatz!
-
Von manchen CYPs weiß man, dass sich ihr Expressionsniveau mit dem Lebensalter ändert. Säuglinge sind z. B. sehr empfindlich auf mit der Muttermilch aufgenommenes Koffein, da sie es selbst nicht abbauen können. Für mehr Ruhe wäre gestressten Müttern zu empfehlen, den Kaffeekonsum einmal versuchsweise zurückzuschrauben.
Enzyminduktion. Das Entgiftungssystem ist nicht starr, sondern in der Lage, sinnvoll auf Beanspruchungen zu reagieren. Wir nehmen häufig kleine Mengen von allerlei Giften auf, da wir uns in einer Welt bewegen, in der jeder versucht, seine Haut und seine ökologische Nische gegen andere zu verteidigen. Betrachten wir zwei Beispiele, die sich zwar nicht so leicht in unseren Magen verirren, aber für unser medizinisches Interesse relevant sind:
- Die pazifische Eibe (Taxus brevifolia) schützt sich davor, als Futter benützt zu werden, indem sie eine Substanz synthetisiert, welche die Funktion von Mikrotubuli und damit der Zellteilungsspindel stört. Wir nennen dieses Gift Paclitaxel und verwenden es als Medikament in der Krebstherapie, z. B. gegen Brustkrebs.
- Das im Erdboden lebende Bakterium Amycolatopsis rifamycinica hält sich dort Konkurrenz vom Leib, indem es eine Substanz synthetisiert, welche die Funktion der DNA-abhängigen RNA-Polymerase anderer Bakterien stört. Wir nennen diese Substanz Rifampicin und verwenden sie als Antibiotikum gegen bakterielle Infektionen, z. B. gegen Meningokokken oder Tuberkulose.
Einschub: Xenohormone (endocrine disruptors). Wir alle gemeinsam beteiligen uns zur Zeit an einem groß angelegten Nahrungsmittelexperiment: Wir nehmen Substanzen auf, mit denen die Menschheit bisher nie konfrontiert war. Nach der Paracelsus-Erkenntnis dosis facit venenum hoffen wir, dass, solange wir nur die Dosis klein halten, nichts passiert. In die Suppe spucken könnten uns dabei Substanzen, die in geringen Konzentrationen wirken, indem sie an hochaffine Rezeptoren binden. In Diskussion sind vor allem lipophile Substanzen, die leicht resorbiert werden, sich oft in Fettgewebe anreichern und an nuclear receptors binden. Wir exprimieren 48 verschiedene nuclear receptors; bei der Mehrzahl von ihnen ist der natürliche Ligand noch unbekannt. Da viele Kernrezeptoren in fast allen Körperzellen exprimiert werden und jeweils auf die Expression hunderter Gene wirken, ist eine Folgenabschätzung von theoretischer Seite unmöglich. Denkbar sind zeitliche Fenster besonderer Empfindlichkeit (z. B. Embryogenese) und epigenetische Effekte auf spätere Generationen. Einige Beispiele aus einer langen Liste von diskutierten Substanzen:
-
Bisphenol A (BPA) hat eine schwache Östrogenwirkung und weitere, unzureichend verstandene Wirkungen. Es wurde z.B. eine Zunahme von T-Helferzellen in der Milz beobachtet, die Auswirkungen auf die Häufigkeit von Allergien haben könnte. Es kommt in Kunststoff-Getränkeflaschen, der Auskleidung von Konserven- und Getränkedosen, Plastikspielzeug und kam bis 2011 in Babyflaschen, bis 2020 auf Thermopapier von Kassazetteln vor. Im Jahr 2023 empfahl die European Food Safety Administration aufgrund neuer Daten eine Reduktion des BPA-TDI (tolerable daily intake) um den Faktor 20.000 (von 4 Mikrogramm/kg Körpergewicht und Tag auf 0,2 Nanogramm).
-
Phthalate haben eine Östrogenwirkung. Sie werden als Weichmacher in PVC, auch in medizinischen Leitungen und Schläuchen, sowie in Kosmetika eingesetzt.
-
Atrazin verändert die ovarielle Hormonproduktion. Es wird in den USA verbreitet als Herbizid eingesetzt, in der EU ist es seit 2003 verboten.
-
Perfluorooctansäure (PFOA) bindet an Östrogenrezeptor und PPARα. PFOA wird wegen ihrer öl- und wasserabstoßenden Eigenschaften zur Herstellung vieler Kunststoffe verwendet, z. B. für Antihaftbeschichtung von Bratpfannen und für Outdoorbekleidung. Sie ist praktisch nicht abbaubar, reichert sich im Fettgewebe an und ist heute in beinahe jedem Individuum nachweisbar. In der EU ist sie seit 2020 verboten.
*
Das System der Biotransformation birgt auch Risiken in sich: eine an sich harmlose Substanz kann durch Biotransformation erst zu einem Toxin werden. Ein klassisches Beispiel dafür ist Aflatoxin B1. Aflatoxin stammt aus Aspergillus flavus, der schlecht gelagerte Erdnüsse, Pistazien, Mais etc. kontaminiert. Das vom Pilz produzierte Molekül ist zunächst inaktiv, wird mit der Nahrung aufgenommen und erst in der Leber durch das Cytochrom-P450-System zu einem hochreaktiven Metaboliten verändert, sodass es DNA-Addukte bildet und damit krebserregend wirkt.
Betrachten wir die Wirkung dieses Systems auf Medikamente etwas näher. Einerseits verlieren Medikamente durch Verstoffwechselung an Aktivität. Bei der Gabe vieler oraler Medikamente tritt der so genannte first pass effect auf: manche Medikamente werden so effizient von der Leber aus dem Pfortaderblut extrahiert und verstoffwechselt, dass es schwierig ist, auf diesem Weg ausreichende Plasmakonzentrationen zu erreichen.
Andererseits kann sich auch bei Medikamenten Toxizität durch Verstoffwechselung ergeben, die wieder durch den Abbau anderer Moleküle beeinflusst werden kann. Ein medizinisch relevantes Beispiel für eine solche Wechselwirkung stellt die Metabolisierung von Alkohol und Paracetamol dar.
1. Alkohol: Ethanol wird hauptsächlich durch das Enzym Alkoholdehydrogenase (ADH) zu Acetaldehyd oxidiert. Bei chronischem Alkoholkonsum wird außerdem CYP2E1 induziert, das denselben Effekt hat. Trotz kräftiger Induktion ist dessen Kapazität jedoch im Verhältnis zur ADH so gering, das es zu keiner substantiellen Steigerung der Alkohol-Abbaurate (0,11 bis 0,12 g/kg Körpergewicht und Stunde, d. h. ca. 0,1 ‰ pro Stunde) kommt. Acetaldehyd wirkt in den so erreichten Konzentrationen bereits zytotoxisch; es wird durch das Enzym Aldehyd-Dehydrogenase weiter zu Azetat oxidiert, anschließend zu Acetyl-CoA aktiviert. In beiden Oxidationsschritten entsteht NADH + H+. Die durch den Alkoholabbau entstehenden Produkte Acetyl-CoA und NADH werden über Zitratzyklus und Atmungskette zur Energiegewinnung eingesetzt; Überschüsse sind das ideale Ausgangsmaterial für die Fettsäuresynthese. Wenn wir Alkohol trinken, synthetisieren wir Fett, statt es abzubauen. Die Verstoffwechselung erklärt die beiden typischen Schädigungsformen durch Alkoholkonsum: alkoholische Hepatitis und Fettleber, die bei fortgesetztem Konsum beide in eine Zirrhose münden können. Ein Überschuss an NADH hemmt nicht nur die β-Oxidation von Fettsäuren. Wie wir bei Metformin gesehen haben, hemmt hohes NADH auch die Gluconeogenese, indem es die Oxidation von Laktat zu Pyruvat hemmt. Die NADH-Konzentration treibt die Reaktion sogar in die Gegenrichtung. Auf diese Weise kann ein Übermaß an Alkohol zu Hypoglykämie und Laktatazidose führen. Typ 2-Diabetiker, die am Vorabend Alkohol getrunken haben, wundern sich oft über ihre "guten" Blutzuckerwerte am folgenden Morgen.
Bei Menschen aus Südostasien kommen zwei Allele häufig vor, die zu einer raschen Akkumulation von Acetaldehyd nach Alkoholkonsum führen. Das ADH-Allel ADH1B*Arg47His führt zu einem rascheren Abbau von Ethanol zu Acetaldehyd, das Aldehyd-Dehydrogenase-Allel ALDH2*2- (Glu504Lys), zu einem verlangsamten Abbau von Acetaldehyd zu Azetat. In beiden Fällen führt Alkoholgenuss rasch zu einer unangenehmen flush-Symptomatik (Gesichtsrötung, Blutdruckabfall), Übelkeit und Kopfschmerzen. In Europa sind die genetischen Grundlagen des Alkoholabbaus sehr homogen.
Obwohl Frauen und Männer Alkohol, bezogen auf das Körpergewicht, in der Leber mit derselben Geschwindigkeit abbauen, ist der weibliche Organismus empfindlicher: Die Aufnahme derselben Menge Alkohol führt bei Frauen zu einer höheren Blutalkoholkonzentration. Das ist nicht nur auf ihr im Schnitt niedrigeres Gewicht zurückzuführen: Alkohol verteilt sich vorwiegend in der wässrigen Phase des Körpers, deren Anteil am Körpergewicht bei Frauen kleiner ist als bei Männern. Ein weiterer Unterschied liegt in einem ADH-Isoenzym, das in der Magenschleimhaut exprimiert wird und bei Frauen weniger aktiv ist. Da mindestens 20 % des getrunkenen Ethanols über die Magenschleimhaut aufgenommen wird, erreicht bei Frauen ein größerer Anteil dieser Fraktion das Blut als bei Männern. Statistisch findet man bei Frauen ab 20 g täglichen Alkoholkonsums einen Anstieg der Zirrhosehäufigkeit, bei Männern erst ab 40-50 g/Tag. Bereits 70-80 g/Tag führen auch bei Männern regelmäßig zur Leberzirrhose.
Die Angabe des Alkoholgehalts von Getränken erfolgt nicht in g, sondern in Volumenprozent (Bier um 5%, also 50 ml pro Liter; Wein um 12%, also 120 ml pro Liter). Da Alkohol leichter ist als Wasser, müssen die ermittelten ml noch mit der Dichte von Alkohol (ca. 0,8 g/ml) multipliziert werden, um die Alkoholmenge in g zu erhalten. 0,5 l Bier enthalten daher ca. 20 g Alkohol, 0,25 l Wein ca. 24 g- damit liegen Tagesmengen von 4 Bier oder einer Bouteille Wein auch bei Männern jenseits der Zirrhosegrenze.
Möchte man nach Alkoholaufnahme die
Alkoholkonzentration im Blut schätzen, so kann man diese in einer ersten
Annäherung nach Widmark berechnen: die Blutalkoholkonzentration ist gleich der
Menge aufgenommenen Alkohols (in g) gebrochen durch das Produkt des Gewichts
der Person (in kg) mal dem Anteil der wässrigen Phase (etwa 0,7 für Männer und
0,6 für Frauen). Dadurch bekommt man g Alkohol pro kg Verteilungsvolumen und
damit
eine Schätzung für die Blutalkoholkonzentration in Promille (g Alkohol pro 1000
g Blut). Diese Schätzung ist in der Regel 10-30% zu hoch, da ein Teil des Alkohols
schon während der Schleimhautpassage verstoffwechselt oder noch während der Trink-
und Resorptionsphase in der Leber verstoffwechselt oder wieder ausgeschieden
wird (Urin, Atemluft). Alternative Berechnungsweisen beziehen
zusätzliche individuelle Variablen wie das Verhältnis Körpergröße/
Körpergewicht und Alter mit ein und liefern genauere Resultate. Möchte man die
Alkoholkonzentration einige Stunden nach Abschluss des Trinkens schätzen, zieht
man vom Ausgangspromillewert die vorher erwähnten 0,11 bis 0,12 ‰ pro Stunde
ab.
2. Paracetamol (im angelsächsischen
Sprachraum als Acetaminophen bezeichnet):
Im therapeutischen Dosisbereich wird der Großteil des Paracetamols
in der Leber direkt sulfatiert und glucuronidiert; nur geringe Mengen
werden über CYP2E1 zu einem hochreaktiven elektrophilen Zwischenprodukt
namens NAPQI (N-Azetyl-p-Benzo-Quinon-Imin) verstoffwechselt, das kovalent
an zelluläre Makromoleküle binden und damit toxisch wirken
könnte. Um reaktive Moleküle dieses Typs abzufangen, produziert
jede Zelle eine gewisse Menge eines SH-tragenden Moleküls, Glutathion.
Das Schwefelatom mit seinen freien Elektronenpaaren reagiert sehr effizient
mit NAPQI und ähnlichen Molekülen; Glutathion stellt damit
einen Schutzschild für die zellulären Makromoleküle dar.
Im toxischen Dosisbereich werden zunächst die Sulfatierungs- und Glucuronidierungssysteme überlastet, so dass nun der zu NAPQI verarbeitete Anteil des verstoffwechselten Paracetamols stark zunimmt. Dann wird das vorhandene Glutathion verbraucht; weiteres NAPQI wirkt ungehemmt toxisch. Wird CYP2E1 durch chronischen Alkoholgenuss oder Medikamente induziert, ist der zu NAPQI verarbeitete Anteil von vornherein größer; Paracetamol wirkt unter diesen Umständen schon in wesentlich niedrigerer Dosis (beschrieben wurden Fälle ab 5-6 Tabletten zu 500 mg pro Tag) toxisch.
Nach diesen Überlegungen wird das Problem für die Pharmakotherapie klar: wenn die wirksame Dosis und Toxizität vieler Medikamente durch CYPs beeinflusst wird, die CYP-Ausstattung einzelner Individuen aber verschieden ist, müssen viele Medikamente in verschiedenen Individuen verschieden wirken. Dafür gibt es zahlreiche Beispiele. CYP2D6 ist wesentlich für den Abbau von Antidepressiva, Neuroleptika und mancher Betablocker, aber auch für die Aktivierung des Opiats Tramadol (Tramal®-Tropfen) zu seiner wirksamen Form. CYP2D6 kommt in vielen Allelvarianten vor: manche Individuen tragen defekte Allele, andere, besonders Menschen aus Äthiopien oder Saudi-Arabien, haben Allele mit mehreren 2D6-Kopien. Für Menschen mit hoher 2D6-Aktivität wirken daher die üblichen Dosen vieler Medikamente gegen Depression oder Schizophrenie nicht, da diese rasch abgebaut werden. Bei Menschen mit fehlender oder niedriger 2D6-Aktivität können dieselben Dosen von Psychopharmaka oder Betablockern toxisch wirken, dafür bleiben Tramadol-Tropfen unwirksam. Ein anderes Beispiel ist der Plättchenhemmer Clopidogrel, der erst durch CYP2C19 in seinen aktiven Metaboliten übergeführt wird. In 10-15% ist CYP2C19 auf Grund allelischer Varianten weniger aktiv, sodass keine ausreichende Plättchenhemmung zustande kommt.
Grundsätzlich ist es möglich, solche potentiell gefährdeten Patienten rechtzeitig zu erkennen, indem man die Gene dieser CYPs auf das Vorhandensein kritischer Polymorphismen überprüft. Ein erster, auf Oligonukleotid-DNA-Chip-Technologie beruhender diagnostischer Test wurde im Jahr 2004 eingeführt. Die immer effizienter werdenden DNA-Sequenzierungs-Verfahren sollten mit der Zeit die Bestimmung eines individuellen Cytochrom P450-Status erleichtern.
Die Möglichkeiten für Komplikationen werden noch dadurch gesteigert, dass sich auf diese genetischen Unterschiede der Cytochrom-P450-Oxidasen Effekte wie Enzym-Induktion, geschlechts- und altersbedingte Enzym-Aktivitätsunterschiede und kompetitive Hemmung bei der gleichzeitigen Einnahme mehrerer Medikamente oder spezifischer Nahrungsinhaltsstoffe aufpfropfen. So hemmen Naringenin und Bergamottin aus Grapefruitsaft CYP3A4 und weitere CYPs und erhöht dadurch die Bioverfügbarkeit zahlreicher Medikamente, z. B. von Statinen.
Funktion: Steroidhormon-Inaktivierung.
Störungen: Gynäkomastie, Hodenatrophie, weiblicher Behaarungstyp.
Das Prinzip, auszuscheidende lipophile Substanzen mit hydrophilen Verbindungen zu konjugieren, um sie wasserlöslicher zu machen, wird auch auf körpereigene Moleküle angewendet, wie z. B. auf Steroidhormone oder Bilirubin. Steroidhormone werden auf diese Weise inaktiviert und ausgeschieden. Chronische Leberinsuffizienz führt beim Mann dazu, dass die in niedrigen Mengen gebildeten Östrogene nicht mehr inaktiviert und ausgeschieden, und damit angereichert werden.
Funktion: Bilirubin-Ausscheidung.
Störung: Ikterus.
Bilirubin ist ein Abbauprodukt von Porphyrinen. Es stammt überwiegend aus der Häm-Gruppe des Hämoglobins und zu einem kleinen Anteil aus der prosthetischen Gruppe von Enzymen der Atmungskette und anderen Enzymsystemen wie z. B. dem Cytochrom-P450-System. Bilirubin wirkt in höheren Konzentrationen toxisch und muss daher vom Körper effizient eliminiert werden. Bilirubin wird über mehrere Transportsysteme in die Hepatozyten aufgenommen, sodass bei diesem Schritt selten Störungen auftreten: organic anion transporter proteins (OATP, in erster Linie OATP1B1, Genname SLCO1B1). Das Enzym UDP-Glucuronyltransferase (UGT) konjugiert Bilirubin mit Glucuronsäure; das konjugierte Bilirubin wird unter ATP-Verbrauch durch den canalicular multispecific organic anion transporter (cMOAT, auch MRP2= mdr related protein 2 genannt, sytematische Bezeichnung ABCC2) in den Gallecanaliculus gepumpt. Bei einer nach der UGT lokalisierten Transportstörung steigt neben dem unkonjugierten Bilirubin auch das konjugierte an, das auch im Urin nachweisbar wird.
Genetisch bedingte Störungen in diesem System sind Gilbert-Meulengracht- (sehr häufig und harmlos, schwächere Funktion des UGT-Promotors), Crigler-Najjar I- und II- (Funktionseinschränkungen unterschiedlicher Intensität der UGT) sowie Dubin-Johnson- (Defekt in cMOAT) und Rotor-Syndrom (äußerst selten; kombinierte Störung zweier OATP, welche die Wiederaufnahme von "entkommenem" konjugiertem Bilirubin aus dem Blut in die Zelle vermitteln).
Funktion: Cholesterin-Ausscheidung.
Störung: Hypercholesterinämie, Dyslipoproteinämien
Cholesterin kann vom Körper synthetisiert, nicht aber wieder abgebaut werden. Es muss also ausgeschieden werden. Dies geschieht in zwei Formen: Cholesterin wird einerseits zu Gallensäuren umgewandelt und andererseits direkt ausgeschieden. 30-60% des in die Galle sezernierten Cholesterins werden im Darm rückresorbiert; der Rest wird ausgeschieden. Bei Cholestase kann ein cholesterinreiches atypisches Lipoprotein entstehen, das Lipoprotein X.
Funktion: (Gallesekretion - in Klammer: Mittel, kein
Ziel).
Störung: Cholestase, Cholelithiasis.
Ein Sistieren des Galleflusses wird unabhängig von der Ursache als Cholestase bezeichnet. Durch die komplexe Zusammensetzung und die für einzelne Komponenten unterschiedlichen Transportsysteme der Galle wird der Begriff Cholestase sowohl für Situationen verwendet, bei denen alle Komponenten der Galle betroffen sind, als auch für Transportstörungen, die hauptsächlich Gallensäuren betreffen. Leitsymptom ist der durch den Anstieg der Gallensäuren ausgelöste Juckreiz.
Gallensäuren werden in Hepatozyten aus Cholesterol synthetisiert, indem Hydroxylgruppen an Position 7 und 12 eingeführt werden und die Cholesterolseitenkette verkürzt und mit einer COOH-Gruppe versehen wird. Die Produktionsrate wird über eine negative Rückkoppelung gesteuert: das geschwindigkeitsbestimmende Enzym, die 7α-Hydroxylase, wird weniger stark exprimiert, wenn genügend Gallensäuren vorhanden sind. Gallensäuren im Hepatozyten binden an den Farnesoid-X-Rezeptor (FXR), ein weiteres Mitglied der Kernrezeptorfamilie, der die Expression des 7α-Hydroxylase-Gens (CYP7A1) unterdrückt. Die neu synthetisierten primären Gallensäuren Cholsäure und Chenodesoxycholsäure werden häufig konjugiert; dafür kommen Taurin, Glycin, Sulfat oder Glucuronat in Frage. Im Darm entfernen Bakterien die 7α-Hydroxylgruppe zum Teil wieder, sodass die sekundären Gallensäuren Desoxycholsäure und Lithocholsäure entstehen. Unkonjugierte Gallensäuren sind sehr schwache Säuren; konjugierte Gallensäuren haben niedrigere pKa und liegen großteils ionisiert als "Gallensalze" vor. Gallensäuren unterliegen einem enterohepatischen Kreislauf: der gesamte Pool aus Gallensalzen und –Säuren wird 5-10 mal am Tag umgewälzt. Durch den enterohepatischen Kreislauf werden auf beiden Seiten des Hepatozyten leistungsfähige Transportsysteme benötigt, die durch die beschriebene Vielfalt der Gallensäuren nicht sehr spezifisch sein können und dadurch auch viele andere Moleküle, z. B. Medikamente, transportieren. Aus dem Pfortaderblut geschieht der Transport hauptsächlich über ein Na+-getriebenes Na‑taurocholate cotransporting polypeptide (NTCP, SLC10A1). Zusätzlich schleust eine Familie von organic anion transport proteins (OATPs, kodiert durch die Gene SLCO1A2, SLCO1B1 und SLCO1B3) ionisierte Gallensalze ein (OATPs schleusen auch das Knollenblätterpilzgift Amanitin in Hepatozyten ein). Gallensäuren in protonierter Form gelangen auch durch non-ionic diffusion in die Hepatozyten. Im Hepatozyten werden Gallensäuren an Proteine gebunden und transportiert. Die Sekretion in den Canaliculus erfolgt unter ATP-Verbrauch hauptsächlich durch die bile salt export pump (BSEP, ABCB11) gegen einen 100- bis 1000-fachen Konzentrationsgradienten; auch cMOAT wird für sulfatierte und glucuronidierte Gallensäuren verwendet. Steigt die Gallensäurenkonzentration im Hepatozyten an, wird über den Farnesoid-X-Rezeptor auch BSEP induziert, sodass mehr Gallensäuren sezerniert werden. Defektallele der BSEP führen zu familiären Cholestasen unterschiedlicher Intensität.
Pharmakologische Querverstrebung: OATP1B1 (Gen: SLCO1B1) transportiert auch Statine aus dem Blut in die Hepatozyten, wo diese einerseits wirken, andererseits metabolisiert und ausgeschieden werden. Ein häufiges – 18-28 % heterozygot, 2-3 % homozygot bei europäischem genetischem Hintergrund – Allel, T521C mit dem Aminosäureaustausch Valin 174 zu Alanin, zeigt eine stark verminderte Transportrate für Statine, sodass die Statin-Plasmakonzentration bei diesen Individuen erhöht ist, was mit einem erhöhten Risiko für Myopathie einhergeht. Dieser Effekt ist nicht für alle Statine gleich; für Simvastatin ist er stärker ausgeprägt als für andere. Andere Medikamente, welche über OATP1B1 aufgenommen werden, verstärken diesen Effekt; dazu gehören Amiodaron und Cyclosporin.
Zusätzlich zu den damit verbundenen Störungen
wie Gallensäurenanstieg
(Juckreiz), Störung der Fettverdauung, Cholesterinanstieg, Ikterus
etc. wirkt eine Cholestase auch negativ auf die Leberzellen zurück.
Gallensäuren sind an sich schon relativ toxische Moleküle.
Durch den Anstieg ihrer Konzentration in den Hepatozyten entstehen
auch atypische, fetale Gallensäuren, die noch toxischer wirken
und die Cholestase verstärken. Cholestase kann viele Ursachen
haben: sie ist eine logische Begleiterscheinung einer schweren Hepatitis,
kann relativ isoliert als Nebenwirkung vieler Medikamente auftreten
(z. B. durch kompetitive Hemmung der BSEP durch Steroide, Ciclosporin
A, Rifampicin) oder kann rein mechanisch durch Gallensteine oder Tumoren
bedingt sein.
Die Entstehung von Gallensteinen ist nicht verwunderlich, wenn man bedenkt, dass die Gallenflüssigkeit an sich bereits einen heiklen Balanceakt zwischen lipophilen Substanzen und ihrem wässrigen Transportmedium darstellt. Typische Größenordnungen der nichtwässrigen Gallebestandteile wären etwa 67% Gallensäuren/-salze und 22% Phospholipide, die benötigt werden, um 4% Cholesterin und Bruchteile eines Prozents Bilirubin-Diglucuronid in Lösung zu halten. Überschreiten die lipophilen Substanzen Cholesterin oder Bilirubin einen gewissen Schwellenwert, sind Gallensäuren und Phospholipide nicht mehr in der Lage, sie in Mizellenform zu halten. Kleine Kristalle können noch in den Darm transportiert werden; gefährlich sind etwas größere Konglomerate, die in den Engstellen stecken bleiben. Große Gallensteine können die Gallenblase vollständig ausfüllen ohne Krankheitserscheinungen auszulösen. Am häufigsten sind Cholesterinsteine; steht Ca-Bilirubinat im Vordergrund, spricht man von Pigmentsteinen.
Funktion: Fettverdauung
Störung: Steatorrhoe, Vitaminmangel ADEK
Galle ist die "Seife", die zur Emulgierung von Nahrungsfetten notwendig ist. Die dabei aktiven Substanzen sind Gallensäuren und Phospholipide. Nahrungsfette sind zu über 90% Triglyzeride, die bei Körpertemperatur flüssig als relativ große Öltropfen vorliegen. Lipasen, die als Proteine eher wasserlöslich sind, können nur an der Wasser-Öl-Grenzfläche aktiv werden. Daher ist für eine effiziente Fettverdauung ein massive Vergrößerung dieser Wasser-Öl-Grenzfläche nötig, die nur durch Zuführen großer Mengen oberflächenaktiver Gallensäuren und Phospholipide erreicht werden kann. Quantitativ überwiegen die biliären Fette im Darm die Nahrungsfette um das Zwei- bis Vierfache. Durch die Wirkung der verschiedenen Lipasen werden neutralere Lipide wie Triglyzeride, Cholesterinester oder Lezithin in kleinere, relativ wasserlöslichere Bruchstücke wie Fettsäuren, Monoglyceride, Cholesterol oder Lysolezithin aufgespaltet, die sich ebenfalls an der Wasser-Öl-Grenzfläche konzentrieren und diese weiter vergrößern. Durch die kontinuierliche Verschiebung zugunsten oberflächenaktiver Moleküle werden die Emulsionströpfchen immer kleiner: von multilamellären über unilamelläre Vesikel (mit Lipiddoppelmembranen) zu gemischten Mizellen, die so klein sind, dass sie nur mehr eine Einzelschicht oberlächenaktiver Moleküle ohne nennenswerten Inhalt darstellen. Diese winzigen Gebilde können nun in die Schleimschicht der Enterozyten diffundieren, die durch einen Na+-getriebenen Protonenantiporter angesäuert wird (der Dünndarminhalt ist durch das Pankreas-Sekret alkalisch). Im sauren Milieu werden die bis dahin ionisierten Fettsäuren protoniert und können durch non-ionic diffusion leichter in den Enterozyten gelangen. Die Aufnahme von Lipidbestandteilen wird auch durch Transportproteine erleichtert. Langkettige Fettsäuren werden z. B. über CD36 aufgenommen. In der Zelle werden die Lipide wieder zusammengesetzt und basolateral als Chylomikronen in die Gewebslymphe freigesetzt. Von dort erreichen sie das Blut im Venenwinkel über die Lymphwege unter Umgehung der Leber. Wird zu wenig Galle in den Darm ausgeschüttet, können Nahrungsfette nur unzulänglich resorbiert werden; der Großteil der Nahrungsfette wird in hellen, voluminösen "Fettstühlen" (Steatorrhoe) wieder ausgeschieden.
Hält dieser Zustand länger an, kann es zu einem Mangel an fettlöslichen Vitaminen kommen. Insbesondere der Mangel an Vitamin K kann zu einer Verstärkung der bei einer chronischen Leberfunktionsstörung ohnehin auftretenden Gerinnungsstörung führen (siehe nächsten Abschnitt). Verglichen mit Vitamin K sind die weiteren fettlöslichen Vitamine von geringerer klinischer Relevanz. Vitamin A wird hauptsächlich in Ito–Zellen gespeichert und gebunden an das auch in der Leber synthetisierte Retinol-bindende Protein im Blut transportiert; sein Mangel macht sich durch verschlechtertes Sehen bei Dunkelheit bemerkbar. Vitamin D wird entweder als Vorstufe mit der Nahrung aufgenommen, oder die Vorstufe wird durch Aufbrechen des Cholesterol-Grundgerüsts durch UV-Einwirkung in der Haut gebildet. In beiden Fällen muss diese Vorstufe in Hepatozyten durch ein Cytochrom P450-Enzym an der Position 25 hydroxyliert werden. Das ist der erste notwendige Schritt, um es in seine aktive Form 1,25-Dihydroxycholecalciferol umzuwandeln, gefolgt von einer zweiten, parathormonregulierten Hydroxylierung in der Niere an Position 1. Vitamin-D-Mangel vemindert die Ca2+-Reserven des Körpers und damit die Knochenmineralisierung. Für das Antioxidans Vitamin E sind keine klar umschriebenen Mangelerscheinungen bekannt.
Pharmakologische Querverstrebung:
·
Das
Transmembranprotein NPC1L1 (Niemann-Pick
C1-Like 1) ist am Transport von Cholesterol aus dem Darmlumen in die
Enterozyten beteiligt. Ezetimib kann
diesen Transport blockieren, sodass einerseits Nahrungscholesterol und
andererseits mit der Galle sezerniertes Cholesterol an der Aufnahme gehindert
wird. Es bewirkt damit eine Absenkung des LDL-Cholesterolspiegels, die jedoch
schwächer als jene durch Statine ist.
· Orlistat blockiert Lipasen im gastrointestinalen Lumen kovalent und damit irreversibel. Es ist in der EU zur Gewichtsreduktion zugelassen. Ungespaltene Fette können nicht resorbiert werden. Die Behandlung erzwingt Nahrungmitteldisziplin, da bereits eine relativ bescheidener Fettgehalt der Nahrung zu explosiven Diarrhöen mit Fettstühlen führt.
Funktion: Plasmaproteinsynthese (Albumin, Gerinnungsfaktoren, Akutphaseproteine, Transferrin, etc.)
Störung: -Hypoproteinämie/Ödeme
-Gerinnungsstörungen
Der Großteil der Plasmaproteine wird von der Leber synthetisiert und sezerniert. Chronische Leberfunktionsstörungen führen daher zu einem Abfall der Plasmaproteinkonzentrationen. Albumin, das mengenmäßig etwa 60% der Plasmaproteine ausmacht, ist über seinen Beitrag zum onkotischen Druck wesentlich dafür, im venösen Schenkel von Kapillarsystemen interstitielle Flüssigkeit rückzuresorbieren und damit das Blutvolumen aufrecht zu erhalten. Der zu niedrige onkotische Druck führt zur Einlagerung von Wasser im interstitiellen Gewebe. Da eine chronische Leberinsuffizienz häufig mit Zirrhose und portaler Hypertension einhergeht, bildet sich durch die Kombination von erniedrigtem onkotischen Druck und erhöhtem Filtrationsdruck häufig ausgeprägter Aszites.
Akutphasenproteinewie C-reaktives Protein (CRP) oder mannanbindendes Protein (MBL), die zur Infektbekämpfung beitragen, werden im Immunologieskriptum besprochen.
Gerinnungsfaktoren sind bei Leberinsuffizienz von zwei Seiten her negativ betroffen: zur allgemein herabgesetzten Syntheseleistung der Leber kommt noch der sekundäre Vitamin K-Mangel. Vitamin K wird benötigt, um eine zusätzliche Carboxylgruppe an Glutaminsäurereste der Faktoren II, VII, IX, X zu hängen. Diese COO−-Gruppen werden benötigt, damit die Gerinnungsfaktoren über Ca2+ an die Phospholipide der Membran aktivierter Thrombozyten binden können (eine gute Methode, um das Gerinnen einer Blutprobe zu verhindern, ist es, Ca2+ durch Zitrat oder EDTA zu binden). Fehlen diese COO−-Gruppen, ist die biologische Aktivität dieser Gerinnungsfaktoren stark herabgesetzt. Vitamin K wird auch im Knochen zur Synthese von Ca2+-bindenden Proteinen wie Osteocalcin benötigt. Symptome durch Vitamin-K-Mangel machen sich natürlich weit früher in der Blutgerinnung als im Knochen bemerkbar.
Pharmakologische Querverstrebung: Cumarinderivate (Acenocumarol, Phenprocoumon) sind Vitamin-K-Antagonisten. Mittels dieser Medikamente wird also ein künstlicher Vitamin K-Mangel erzeugt, um die Gerinnbarkeit des Blutes herabzusetzen (z. B. nach einer Lungenembolie). Durch den biologischen Mechanismus wird auch klar, dass es nach Absetzen dieser Medikamente, z. B. für einen zahnmedizinischen Eingriff, relativ lange dauert, bis funktionierende Gerinnungsfaktoren nachsynthetisiert werden können.
Funktion: Monitoring des Einstroms vom Darm her in einem Niederdruckkapillarsystem
Störung: Portale Hypertension
Alle Einwirkungen, die zu einem ausgeprägten Absterben von Leberzellen führen (z. B. Virushepatitis, Alkohol, anhaltende Cholestasen) führen über Regenerationsversuche auch zu einem sekundären Umbau der Leber. Zusätzlich werden die Sternzellen (Ito-Zellen) aktiviert, verstärkt extrazelluläre Matrix in der Form von Kollagen und Proteoglykanen zu bilden: es entsteht eine Leberfibrose. Der Stoffaustausch zwischen Blut und Hepatozyten wird durch eine Abnahme an Endothelzellfenestrationen und verlängerte sowie verdichtete Diffusionsstrecken behindert. Diese Veränderung der Architektur schließt die Gefäße mit ein und resultiert in einer Verringerung des Gesamtquerschnitts aller Pfortaderverästelungen und damit in einem Druckanstieg (man kann sich das bildlich wie eine "Verstopfung des Filters" vorstellen). Folgen dieses Druckanstiegs sind Hypersplenismus, die Ausweitung portocavaler Anastomosen und eine verstärkte Aszitestendenz.
Hypersplenismus bedeutet, dass Blutzellen verstärkt in der Milz zurückgehalten ("sequestriert") werden. Die Milz nimmt dadurch wesentlich an Größe zu; schließlich erfolgt auch ein verstärkter Abbau von Blutkörperchen und Thrombozyten.
Shunts über portocavale Anastomosen bedeuten, dass das vom Darm kommende Blut an der Leber vorbei ("ungefiltert") in den großen Kreislauf gelangt. Zu den dadurch verstärkten Erscheinungen der Leberinsuffizienz kommt noch die Gefahr schwer zu stillender Blutungen aus Ösophagusvarizen.
Beim hepatorenalen Syndrom kommt es durch mehrere Mechanismen – Volumenverlust aus dem Gefäßsystem, arterielle Vasodilatation im Splanchnikusgebiet – zu einer Reduktion des Kreislaufvolumens und damit einer renalen Vasokonstriktion, die zu einer ausgeprägten Funktionseinschränkung der Niere führen kann. Der juxtaglomeruläre Apparat versucht den Rückgang des effektiven Kreislaufvolumens durch Aktivierung des Renin-Angiotensin-Aldosteronsystems zu kompensieren; der sekundäre Hyperaldosteronismus verstärkt seinerseits den Aszites durch Na+- und Wasserretention.
***
QUELLEN UND WEITERFÜHRENDE LITERATUR:
Höfler G. et al. (Hrsg.): Pathologie, 6. Auflage, Urban und Fischer, 2019
Schwarz et al. (Hrsg.): Pathophysiologie, Maudrich, Wien, 2007
Siegenthaler W. und Blum H. E.(Hrsg.):
Klinische Pathophysiologie, 9. Auflage, Thieme, Stuttgart, 2006
auf Englisch:
Kumar V. et al. (eds.): Robbins and Cotran Pathologic Basis of Disease, 10th Edition, Elsevier, 2020
Boron W. F. and Boulpaep E. L. (eds.): Medical Physiology, 3rd Edition, Elsevier, Philadelphia, 2016